

️ Đánh giá tính an toàn của vaccine (P1)
.png)
Sinh viên ĐHYD Châu Hào Nam
Lưu Tuyết Hoa
Đặng Văn Vương
Trần Phương Nam
Lê Trần Phương Uyên
ThS. Đỗ Đăng Trí

Sự quan trọng của việc nghiên cứu an toàn vaccine
Các báo cáo về sự kiện có hại của vaccine bao gồm các sự kiện sức khỏe do vaccine gây ra (tức là phản ứng có hại thực sự) và những trường hợp không phải do vaccine gây ra (chỉ xảy ra do trùng hợp). Trong các chương trình tiêm chủng đang hoàn thiện (Hình 1), giám sát chặt chẽ và đánh giá kịp thời các tác dụng phụ nghi ngờ vaccine là rất quan trọng để ngăn ngừa mất niềm tin, giảm tỷ lệ bao phủ vaccine và dịch bệnh quay trở lại.
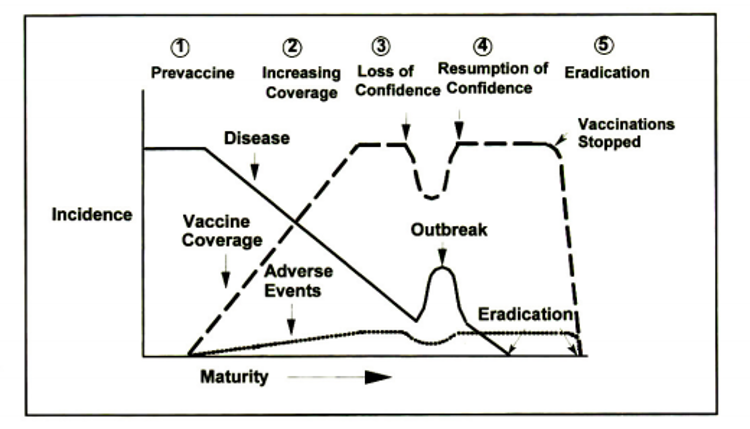
Hình 1. Quá trình phát triển của chương trình tiêm chủng và sự nổi bật về tính an toàn vaccine
Có một số lý do thiết yếu để hỗ trợ và duy trì một chương trình an toàn vaccine đang hoạt động. Mong đợi về tính an toàn của vaccine thường cao hơn của các loại can thiệp y tế khác bởi vì hầu hết các dược phẩm được sử dụng cho người đã có bệnh và vaccine được tiêm cho những người khỏe mạnh. Khả năng chấp nhận của cộng đồng đối với các phản ứng có hại liên quan đến các sản phẩm được sử dụng cho người khỏe mạnh, đặc biệt là trẻ sơ sinh khỏe mạnh, về cơ bản thấp hơn đáng kể so với các sản phẩm được sử dụng cho những người đã bị bệnh. Mức độ chấp nhận rủi ro thấp hơn này của công chúng đối với vaccine dẫn đến nhu cầu cao hơn để nghiên cứu các nguyên nhân hiếm gặp có thể gây ra tác dụng phụ sau khi tiêm vaccine so với hầu hết các sản phẩm dược phẩm khác.
Một lý do khác là dựa trên việc sử dụng rộng rãi vaccine và tác động tiềm ẩn của các vấn đề hoặc mối quan tâm để phát triển trong một tỷ lệ lớn dân số tiếp nhận vaccine.
Không giống như nhiều loại thuốc có thể được thay thế, vaccine nói chung có ít chủng loại hoặc lựa chọn thay thế. Quyết định thu hồi vaccine hoặc chuyển đổi giữa các chủng có thể có sự phân tán rộng rãi (wide ramifications). Ví dụ, việc thu hồi vaccine “cúm lợn” năm 1976 do nguy cơ cao gây hội chứng Guillain-Barré đã dẫn đến nhiều vụ kiện và sự chấp nhận của công chúng đối với việc tiêm phòng cúm trong giai đoạn đó. Việc thiết lập các hiệp hội về vaccine và xác định kịp thời các rủi ro là rất quan trọng trong việc đặt các sự kiện bất lợi vào quan điểm rủi ro-lợi ích thích hợp. Một sự liên kết sai lầm hoặc rủi ro quy kết có thể làm giảm niềm tin vào vaccine và gây ra những hậu quả tai hại cho việc chấp nhận vaccine và tăng tỷ lệ mắc bệnh. Mặt khác, việc phủ nhận sự liên quan giữa sự kiện bất lợi và vaccine mặc dù đã tích lũy được bằng chứng cũng có thể làm suy giảm lòng tin của công chúng.
Nghiên cứu về tính an toàn của vaccine có thể giúp phân biệt các phản ứng vaccine thực sự với các sự kiện ngẫu nhiên không liên quan và có thể giúp duy trì niềm tin của công chúng đối với việc tiêm chủng và độ tin cậy của các chương trình tiêm chủng.
Giai đoạn tiền lâm sàng - Những yếu tố quan trọng
Nhận định chung
Mục đích của những nghiên cứu tiền lâm sàng đối vaccine COVID-19 nhằm nhận định tính sinh miễn dịch và độ an toàn thông qua các thử nghiệm in vitro và invivo. Những thử nghiệm trên động vật có thể hỗ trợ nhận diện những nguy cơ tiềm ẩn và đưa ra hướng dẫn ban đầu về liều tiêm, số lần tiêm và đường tiêm của vaccine khi tiên hành thử nghiệm lâm sàng. Các dữ liệu tiền lâm sàng đầy đủ sẽ thuận lợi hơn cho quá trình tiến hành thử nghiệm trên người (first in human -FIH).
FDA khuyến cáo những nhà sản xuất vaccine nên chủ động liên hệ với FDA ngay từ đầu để bàn luận về những dữ liệu được yêu cầu cho loại vaccine đang phát triển nhằm tạo điều kiện thuận lợi hơn cho những thử nghiệm trên người và các thử nghiệm lâm sàng sau đó.
Các nghiên cứu về độc tính
Đối với loại vaccine COVID-19 đang phát triển là một cộng nghệ mới chưa có các thử nghiệm tiền lâm sàng hay lâm sàng trước đây thì các nghiên cứu về an toàn tiền lâm sàng là bắt buộc trước khi tiến hành thử nghiệm lâm sàng FIH.
Trong một số trường hợp, có thể không cần thực hiện các nghiên cứu an toàn tiền lâm sàng trước khi thử nghiệm lâm sàng FIH vì thông tin sẵn có đủ để mô tả tính an toàn của sản phẩm. Ví dụ: nếu vaccine COVID-19 được sản xuất bằng công nghệ đã được nghiên cứu để sản xuất vaccine trước đó và có đủ yêu cầu cần thiết, thì có thể sử dụng dữ liệu độc chất từ những nghiên cứu này (dữ liệu từ các nghiên cứu độc tính khi lặp lại liều, về biodistribution) và dữ liệu lâm sàng được thu thập từ các sản phẩm khác cùng một nền tảng công nghệ để hỗ trợ các thử nghiệm lâm sàng FIH cho vaccine COVID-19 đó. Các nhà sản xuất vaccine nên tóm tắt các phát hiện quan trọng và cung cấp cơ sở lý luận khi sử dụng những dữ liệu này thay cho việc thực hiện các nghiên cứu an toàn tiền lâm sàng.
Tại Hoa Kỳ, khi cần hỗ trợ để tiến hành thử nghiệm lâm sàng FIH, các đánh giá độ an toàn tiền lâm sàng bao gồm độc tính và dung nạp tại chỗ phải được thực hiện đáp ứng được các tiêu chuẩn về Good Laboratory Practices (GLP) trong quy định liên bang (Code of Federal Regulations- CFR) đưa ra. Những nghiêm cứu này nên được thực hiện đầy đủ và hoàn thành trước khi thử nghiệm lâm sàng FIH.
Dữ liệu về độc tính có thể gửi bản thảo trước khi thẩm định chính thức nhằm đẩy nhanh tiến trình thử nghiệm trên người. Bản báo cáo đầy đủ cuối cùng nên được gửi cho FDA trong vòng 120 ngày sau khi bắt đầu thử nghiệm lâm sàng.
Sử dụng vaccine phòng COVID-19 liên quan đến phụ nữ mang thai là một cân nhắc quan trọng đối với các chương trình tiêm chủng. Do đó, FDA khuyến cáo rằng trước khi thực hiện tiêm chủng, các nhà sản xuất nên tiến hành các nghiên cứu về độc tính đối với sự phát triển và sinh sản (Developmental And Reproductive Toxicity – DART) cho vaccine COVID-19 mà họ đang phát triển. Ngoài ra, nhà sản xuất có thể gửi dữ liệu có sẵn các nghiên cứu về DART của một sản phẩm tương tự sử dụng cùng một nền tảng công nghệ kỹ thuật nếu được FDA tham khảo và đồng ý rằng những dữ liệu đó là đầy đủ về mặt khoa học.
Các nghiên cứu về Biodistribution khi thử nghiệm trên động vật nên được xem xét thực hiện nếu thành phần vaccine không có dữ liệu liên quan trước đây.
Đặc tính sinh miễn dịch khi thử nghiệm trên người
Các nghiên cứu về đặc tính sinh miễn dịch trên động vật khi được tiêm vaccine nên được thực hiện nhằm hỗ trợ thử nghiệm lâm sàng FIH.
Các nghiên cứu sẽ đánh giá các phản ứng miễn dịch dịch thể, miễn dịch tế bào và những chức năng miễn dịch khác đối với loại vaccine đang phát triển.
Những nghiên cứu về hiện tượng ERD liên quan đến vaccine (Vaccine-associated Enhanced Respiratory Disease)
ERD đề cập đến những triệu chứng lâm sàng nghiêm trọng sau khi nhiễm virus, như RSV (virus hợp bào hô hấp) virus cúm, do đã có đáp ứng miễn dịch khi tiếp xúc trước đó. ERD thường biểu hiện sự thâm nhiễm bạch cầu ái toan tại phế quản. ERD có thể xảy ra khi nhiễm tác nhân virus cùng loài (homotypic) hay dị loài (heterotypic) sau tiêm vaccine, trong môi trường tự nhiên hoặc miễn dịch thụ động từ mẹ sang con.3
Các dữ liệu nghiên cứu trên mô hình động vật sau khi tiêm một số loại vaccine kháng virus corona trước đây (SARS-CoV và MERS-CoV) làm tăng mối lo ngại về hiện tượng ERD (enhanced respiratory disease) liên quan đến vaccine COVID-19. Trong những nghiên cứu này, động vật thử nghiệm sẽ được tiêm vaccine kháng một loại virus corona và cho nhiễm với tác nhân virus tương ứng ngay sau đó. Những nghiên cứu khi ấy cho thấy tính sinh miễn dịch tại phổi có sự gia tăng hoạt động tế bào Th-2 tương tự như những hiện tượng ERD trước đó được mô tả khi tiêm vaccine formalin-bất hoạt của virus hợp bào hô hấp (RSV) cho trẻ sơ sinh và động vật khi bị nhiễm từ môi trường tự nhiên hay trong phòng thí nghiệm.2 Đã có những cảnh báo trước đó về ERD nên được đánh giá đầy đủ đối với vaccine coronavirus đang thử nghiệm để tránh lặp lại thất bại như những lần trước đó. Vaccine RSV đầu tiên, dựa vào formalin-inactivated RSV (FI-RSV), đã tiến hành thử nghiệm lâm sàng vào năm 1965, thời điểm khi đó đã có một số loại vaccine virus bị bất hoạt hay giảm độc lực được phát triển thành công như đậu mùa, bại liệt. Vaccine FI-SRV ban đầu cho thấy dung nạp tốt và sinh miễn dịch vừa phải. Tuy nhiêm, thay vì có tác dụng phòng ngừa cho những tình nguyện viên, FI-SRV lại gia tăng một hiệu ứng nghịch lý gây ra hiện tượng ERD khi người được tiêm vaccine tiếp xúc tác nhân virus trong môi trường tự nhiên. Trong số 20 đứa trẻ được tiêm ngừa lúc ấy, có đến 16 trẻ được cho nhập viện, bao gồm 2 ca tử vong ngay sau đó. Trong khi chỉ có 1 ca nhập viện bên nhóm chứng. Vì vậy, FDA buộc dừng khẩn cấp thử nghiệm lâm sàng vaccine RSV.3
Những loại vaccine đang thử nghiệm nên được làm sáng tỏ bằng những nghiên cứu về vấn đề này. Những hiểu biết hiện tại về nguy cơ tiềm ẩn của vaccine Covid-19 liên quan đến ERD còn hạn chế, cũng như để hiểu giá trị của thử nghiệm lâm sàng trong việc dự đoán nguy cơ của hiện tượng này trên cơ thể người. Dù vậy, các nghiên cứu trên động vật (chuột, một số loài linh trưởng,…) được cho là quan trọng nhằm tiếp cận ERD liên quan đến vaccine.
Các nghiên cứu về việc gây nhiễm sau tiêm vaccine trên mô hình động vật (Post-vaccination) và đặc tính sinh miễn dịch tiền lâm sàng và lâm sàng do tác nhân Covid-19 nhất định sẽ giúp đánh giá khả năng vaccine đó gây ra hiện tượng ERD ở người hay không.
Trước khi tiến hành thử nghiệm lâm sàng FIH, các nhà sản xuất nên thực hiện các nghiên cứu và đặc tính sinh miễn dịch tạo sau tiêm vaccine trên mô hình động vật nhằm đánh giá các dấu chứng (markers) về nguy cơ gây ERD. Những nghiên cứu này bao gồm đánh giá đặc tính sinh miễn dịch (tỉ lệ kháng thể trung hoà so với kháng thể toàn phần được tạo ra,…), độ chênh lệch về số lượng tế bào Th1/Th2 sau tiêm vaccine COVID-19 ở động vật.
Các vaccine COVID-19 đang phát triển có những dữ liệu liên quan trước đây cho thấy nồng độ kháng thể trung hoà cao và gia tăng hoạt động của tế bào Th1 thì sẽ thực hiện song song thử nghiệm lâm sàng FIH và trên động vật trước khi triển khai các pha 2 và pha 3 nhằm giảm thiểu nguy cơ ERD ở người. Đặc biệt là cần trang bị đầy đủ nhằm xử trí kịp thời các vấn đề liên quan ERD khi thử nghiệm trên người. Ngược lại, nếu vaccine đang phát triển chưa có đầy đủ dữ liệu phù hợp về đặc tính sinh miễn dịch thì nên thử nghiệm trên động vật trước khi tiến hành thử nghiệm lâm sàng FIH.
Toàn bộ dữ liệu liên quan đến một loại vaccine cụ thể, bao gồm vấn đề gây nhiễm sau tiêm vaccine trên động vật và các thử nghiệm lâm sàng FIH có mô tả đặc tính sinh miễn dịch sau tiêm vaccine sẽ được xem xét cẩn thận trước khi tiến hành thử nghiệm lâm sàng ở những giai đoạn sau.2
Các pha nghiên cứu lâm sàng
Không giống như những loại thuốc dùng trên người bệnh, vaccine được tiếp nhận bởi những cá thể có tình trạng sức khoẻ khác nhau; chính vì vậy mà độ an toàn cần phải ở mức cao hơn rất nhiều. Mỗi loại vaccine trước khi được chấp thuận phải trải qua quá trình sàng lọc và đánh giá để xác định kháng nguyên nào nên được sử dụng để kích hoạt đáp ứng miễn dịch. Sau khi trải qua giai đoạn tiền lâm sàng, nếu vaccine đó tạo được đáp ứng miễn dịch trên động vật, sẽ đi tiếp vào 3 pha thử nghiệm lâm sàng trên con người.
Pha I
Ở pha này, mục tiêu chính là đánh giá tính an toàn và khả năng sinh phản ứng, bên cạnh mục tiêu phụ là thu thập đáp ứng miễn dịch. Thông thường, liều lượng, thời gian tạo miễn dịch cũng được đánh giá sơ bộ ở pha này. Vaccine sẽ được tiêm trên một nhóm nhỏ các đối tượng trẻ tuổi, khoẻ mạnh, sức đề kháng tốt, hay nói cách khác là có nguy cơ thấp nhiễm bệnh liên quan tới loại vaccine đang tiêm (phân tích dựa trên huyết thanh học, tiền sử tiếp xúc và phơi nhiễm).
Thử nghiệm ở pha này thường “mở nhãn” và không ngẫu nhiên, nhưng có thể tiến hành các thử nghiệm ngẫu nhiên có đối chứng (RCT – randomized controlled trials), trong đó giả dược hoặc một loại vaccine ngăn ngừa một căn bệnh khác có thể được dùng cho nhóm chứng. Để kiểm soát sự sai lệch và tăng độ chính xác, có thể tiến hành nghiên cứu theo hướng mù đôi hoặc mù đơn. Trong đó, việc thực hành sử dụng công thức trộn giữa kháng nguyên vaccine và chất bổ trợ ngay trước khi chủng ngừa được áp dụng, nhằm cho phép người phát triển vaccine có thể thử nghiệm nhiều loại chất bổ trợ mà không cần quá nhiều công thức vaccine. Các công thức đều được bào chế theo quy trình nhiều lớp, tuy nhiên, bất kì thay đổi nào trong công thức đều sẽ phải được thử nghiệm lại ở một pha I mới hoàn toàn.
Điểm đặc biệt đó là, vaccine sống giảm độc lực/ vaccine từ vi sinh vật chết có thể gây hoang mang, lo ngại vì khả năng phát tán tác nhân lây nhiễm, truyền từ người sang người, và có thể diễn tiến đến một tình trạng độc lực cao hơn. Vì thế, những đối tượng tham gia pha I cần được giám sát chặt chẽ, quan sát và phát hiện kịp thời bất kì dấu hiệu lâm sàng nào của sự nhiễm trùng. Đối tượng được làm những xét nghiệm thường quy (huyết học, sinh hoá, phân tích nước tiểu…) nhằm theo dõi tính an toàn và đồng thời tạo thành dữ liệu ban đầu, với các khoảng cách thời gian xác định và khi kết thúc thử nghiệm.
Pha II
Những đối tượng tham gia nghiên cứu sẽ bước vào pha II sau khi đạt được kết quả khả quan trong pha I cả về tính an toàn lẫn tính sinh miễn dịch. Mục tiêu của pha II là đánh giá liều lượng tối ưu và các điểm cuối về tính an toàn, dung nạp, đáp ứng miễn dịch, hiệu quả và cả những phản ứng phụ tiềm ẩn. Các nghiên cứu này có sức mạnh thống kê và cỡ mẫu xác định, chính vì thế được kì vọng sẽ cung cấp một kết quả có ý nghĩa lâm sàng. Các nghiên cứu giai đoạn này chọn ra khoảng 100-1000 thuộc quần thể “mục tiêu”, họ tin tưởng vào các nhà nghiên cứu rằng vaccine này sẽ có tính bảo vệ và an toàn tương đối. Đối tượng nghiên cứu có thể bao gồm người lớn, thanh thiếu niên, trẻ em, trẻ sơ sinh hoặc thậm chí là phụ nữ có thai. Tuy nhiên, đối với những vaccine dành cho trẻ sơ sinh, phương pháp tiếp cận “từng bước, lần lượt” sẽ được tiến hành, thực hiện tuần tự trên người lớn – thanh thiếu niên – trẻ em – trẻ sơ sinh.
Trong thí nghiệm thiết kế một cách ngẫu nhiên, có đối chứng, vaccine được nghiên cứu đối đầu với giả dược hoặc một loại vaccine khác để xem liệu những thay đổi trong nhóm tiêm chủng là do vaccine hay xuất hiện một cách tình cờ. Các nghiên cứu này nhằm mục đích ghi nhận hiệu quả trên những nơi có tỷ lệ nhiễm bệnh cao để đưa ra kết quả tốt nhất. Ví dụ, hai nghiên cứu pha IIb tương tự đối với vaccine Rotarix ở Mỹ La-tinh và Singapore để đánh giá tính an toàn, sinh miễn dịch và hiệu quả ở các nồng độ/ liều khác nhau. Cả hai nghiên cứu đều có đối chứng với giả dược, tiến hành song song với cỡ mẫu tương đương nhau, và đạt được tỉ lệ chuyển đảo huyết thanh tốt, nhưng nghiên cứu ở Mỹ La-tinh cũng đã chứng minh hiệu quả chống lại 2 týp huyết thanh của virus Rota. Rõ ràng là từ quá trình lâm sàng của các loại vaccine khác nhau, cho thấy sự cần thiết của nhiều nghiên cứu thực hiện ở pha II nhằm giải quyết những ảnh hưởng của biến số về liều lượng, nhóm tuổi, thời gian theo dõi… trước khi tiếp tục nghiên cứu pha III.
Phản ứng miễn dịch dịch thể và miễn dịch tế bào đối với các tác nhân trong vaccine cần được đánh giá trên những đối tượng tiêm. Phương thức bảo vệ của vaccine sẽ quyết định phương pháp đo đạt đáp ứng miễn dịch của cơ thể. Ví dụ, với Rotarix, nồng độ kháng thể IgA huyết thanh được theo dõi, trong khi đối với Gardasil, nồng độ kháng thể IgG huyết thanh được theo dõi để xác định tỷ lệ chuyển đổi huyết thanh. Hiếm khi những nghiên cứu trong pha II được cấp phép rằng những marker miễn dịch này là kết quả sau cùng, như vaccine não mô cầu (MCC) được cấp phép dựa trên các tương quan huyết thanh học và khả năng bảo vệ mà không có dữ liệu về độ hiệu quả, ở Vương quốc Anh.
Tóm lại, pha II sẽ cung cấp những thông tin sơ bộ về hiệu quả bảo vệ thông qua thí nghiệm trên đối tượng khoẻ mạnh đã được cho nhiễm mầm bệnh một cách có chủ ý. Các nghiên cứu như vậy gọi là nghiên cứu giai đoạn IIa, và chỉ thích hợp cho những bệnh được công nhận về mặt khoa học và y đức – nghĩa là mầm bệnh không gây nhiễm trùng dẫn đến tử vong và không kháng với các phương pháp điều trị bệnh sẵn có. Những nghiên cứu mang tính “thử thách” này cũng đã từng thực hiện để kiểm tra hiệu quả ban đầu của sốt rét, thương hàn, dịch tả…
Pha III
Các thử nghiệm then chốt trong pha này nhằm đánh giá hiệu quả của công thức vaccine cuối cùng, trước khi vaccine được đăng ký và chấp thuận trên thị trường. Hiệu quả của vaccine (VE) được định nghĩa là phần trăm số người giảm tỷ lệ nhiễm bệnh trên tổng số người được tiêm chủng. Cho rằng rủi ro tương đối là RR, tỷ lệ mắc bệnh trong số các đối tượng chưa tiêm chủng là Iu và trong số các đối tượng đã tiêm là Iv, VE được tính bằng:
(Iu-Iv/Iu) × 100% = (1- Iv/Iu) × 100% = (1-RR) × 100%
Đích đến cuối cùng trong pha này đó là giảm tần số mắc bệnh, tuy nhiên, nó cũng có thể dựa vào các “đích đến” khác, chẳng hạn như tỷ lệ nhiễm trùng hoặc các tương quan giữa miễn dịch và khả năng bảo vệ cá thể.
Đây là một pha tổ chức với quy mô lớn, tiếp nhận 1.000-10.000 đối tượng từ dân số mục tiêu. Các nghiên cứu thực địa sẽ tương tự như mô hình sử dụng vaccine trong tương lai. RCT được xem là “tiêu chuẩn vàng”, trong đó người tham gia phân bố ở nhiều khu vực trong một quốc gia hoặc nhiều quốc gia khác nhau để nhận vaccine đang điều tra hoặc vaccine đối chứng (giả dược, nước muối, hoặc vaccine khác). Một RCT tốt và tiềm năng sẽ kiểm soát được các biến số, ngăn ngừa sai lệch và tối đa hoá cơ hội phát hiện ra điểm khác biệt giữa vaccine và nhóm chứng. Trong các nghiên cứu pha III, quan trọng nhất là tạo được miễn dịch cộng đồng bên cạnh những khía cạnh về mặt lâm sàng. Việc đánh giá hiệu quả bảo vệ chủ yếu tập trung vào khả năng ngăn ngừa bệnh của vaccine, ngoài ra còn có thể đánh giá một số phương diện khác như:
- Hiệu quả của vaccine đối với tính nhạy cảm, khả năng xâm nhập, phát triển, gây bệnh và lây nhiễm của vi sinh vật.
- Tổng hiệu lực vaccine
- Tác động gián tiếp lên những đối tượng không được tiêm chủng như thế nào?
- Tổng tác dụng của việc tiêm chủng ở những người đã được tiêm chủng
- Ảnh hưởng của vaccine ở cấp độ dân số tổng thể
Nếu kết quả pha III chứng minh được tính hiệu quả và an toàn, nhà sản xuất có thể nộp đơn lên cơ quan quản lý quốc gia để được cấp phép và tiếp thị sản phẩm.
Xem tiếp: Đánh giá tính an toàn của vaccine (P2)
Bệnh viện Nguyễn Tri Phương - Đa khoa Hạng I Thành phố Hồ Chí Minh
BÀI VIẾT KHÁC



