

️ Thay thế các gen kiềm chế khối u trong ung thư (P1)
MỞ ĐẦU
Trước khi cơ sở di truyền của ung thư được chấp thuận và trên một nửa thế kỷ trước khi thuật ngữ gen kiềm chế khối u (tumor suppressor gene - TSG) được đặt tên, nhà sinh học Đức Theodor Boveri đã cho rằng khi kiềm chế các nhiễm sắc thể xác định sẽ làm ức chế sự phân chia tế bào. Theo bản dịch, xuất bản năm 1929 thì ngay từ năm 1914 Boveri đã mạnh dạn viết các tế bào khối u với sự tăng trưởng không giới hạn sẽ xuất hiện nếu loại đi các nhiễm sắc thể bị ức chế. Sau đó hơn một nửa thế kỷ, với việc phân tích phân tử các khối u ở người đã vạch rõ rằng: mỗi trường hợp ung thư đều có ít nhất là một biến đổi di truyền phức hợp trong một nhiễm sắc thể bị ức chế, ngày nay được hiểu chính là gen kìm chế khối u (TSG). Trong những năm 1980, sinh học phân tử và di truyền học ung thư đã chuyển các nST bị ức chế theo thuyết của Boveri thành các công cụ sắc bén cho việc nghiên cứu sinh bệnh học phân tử (molecular pathogenesis) ung thư và tới những năm 1900 thì chúng được đẩy từ phòng thí nghiệm tới tận giường bệnh. Từ giữa 1994 đến cuối 2002 đã có trên 25 thử nghiệm lâm sàng về trị liệu thay thế TSG đã được thực hiện, những kết quả thu được đã vach ra một tương lai đầy hứa hẹn của TSG trong việc quản lý ung thư.
Vì những kiến thức thu được về TSG người ngày càng nhiều nên trong chương này chúng tôi chỉ xin tóm lược về sinh học TSG đồng thời cũng đưa ra một số định hướng chi tiết. Hơn nữa, các dẫn liệu thu được từ các nghiên cứu tiền lâm sàng và lâm sàng ngày càng chồng chất, vì thế trong khi thảo luận về các chiến lược gen trị liệu với việc thay thế TSG chúng tôi chỉ giới hạn ở một trong số các TSG đã bị biến đổi phổ biến nhất, đó là p53.
CÁC GEN KIỀM CHẾ KHỐI U
Sự tiến bộ nhanh chóng trong những lĩnh vực sinh học và di truyền học phân tử đã tạo ra các công cụ cho phép chúng ta can thiệp sâu vào những công việc bên trong các tế bào bình thường cũng như các tế bào ác tính, hay các mạng lưới phức tạp của các con đường hóa sinh. Quá trình thực nghiệm gỡ rối mạng lưới đó có liên quan tới việc chắp lại các đơn vị thông tin thu lượm được từ các dòng tham vấn khác nhau, một quá trình mà đôi khi dường như không nhận được các câu trả lời khẳng định về sinh hóa học ung thư. Watson đã tóm tắt vấn đề này trong chương kiểm soát sự tăng sinh của tế bào (xuất bản năm 1977) với cả sự lạc quan xen lẫn hoài nghi:
“Về vấn đề một số khác biệt có thể được sao chép lại đã nói với chúng ta rằng, các nghiên cứu về ung thư ở mức phân tử không còn là một khoa học viển vông theo kiểu đông - ky- sốt nữa. Mặt khác, chúng ta không được làm cho chính chúng ta phải thất vọng bởi vì chúng ta có thể đo được hàm lượng AMP vòng và nếu thông minh hơn nữa chúng ta sẽ khảo sát GMP vòng. Theo bất kỳ nghĩa nào chúng ta cũng đang tiến gần tới đỉnh của các vấn đề ung thư nhiều công trình nghiên cứu về ung thư vẫn đang tiếp tục tương tự như việc tìm kiếm một đồng xu bị mất ven một con đường mà đôi khi chỉ được rọi sáng bằng ánh đèn chiếu sáng của đường phố. Điều cần thiết là chúng ta phải được nhìn vào những vùng được chiếu sáng, còn nếu chúng ta có nhìn thật lâu đến đâu đi nữa trong bóng tối thì việc tìm kiếm cũng không bao giờ thành công. Vì vậy hầu hết các thành phần của màng tế bào bình thường vẫn là những chiếc hộp đen, và hóa sinh học ung thư vẫn là sự huyền bí đối với các thế hệ tương lai.
Tuy vậy, chỉ vài năm sau đó các kỹ thuật đã được cải tiến cho việc thao tác các phân tử sinh học đã làm lóe sáng trên những chiếc hộp đen đó, dần dần người ta đã khám phá ra các thông tin chi tiết hóa sinh về mạng lưới các con đường chế ngự các quá trình tế bào và phát hiện ra rằng nhiều gen có liên quan tới việc điều hòa quá trình tăng trưởng của các tế bào bình thường (phân chia, biệt hóa và chết của tế bào) đã bị biến đổi ở các tế bào khối u.
Với việc so sánh giữa các tế bào bình thường và các tế bào ác tính ở mức độ gen đã nhanh chóng chỉ ra sự liên can của các gen ung thư trội trong việc suy diễn căn nguyên ung thư, nhưng thông tin về các thành viên của họ TSG thì vẫn còn rất khó hiểu. Mặc dầu đã có bằng chứng sinh học về sự tồn tại của gen kiềm chế khối u từ những công trình trong những năm 1970 và 1980, nhưng các công trình về hóa sinh, sinh học của TSG ở mức phân tử thì vẫn đòi hỏi phải có các chiến lược tiên tiến hơn nữa khi nghiên cứu sinh học gen ung thư và phải làm sáng tỏ vai trò của TSG trong các quá trình ở tế bào bình thường cũng như các tế bào ung thư. Trong những năm 1990, người ta đã phát hiện ra sự mất chức năng của một vài TSG ở khá nhiều dạng ung thư khác nhau, phúc đáp lại các câu hỏi về sinh bệnh học phân tử của ung thư và con đường dẫn tới các mẫu di truyền hiện tại: ung thư là kết quả của sự tích lũy đa tổn thương di truyền trong suốt thời gian sống của một cá thể. Khi mất chức năng ở TSG thì sẽ dẫn tới ung thư. Trong các ung thư có tính chất gia đình thì một alen biến đổi được di truyền lại như một đột biến mầm, còn alen kia là kết quả của sự biến đổi thu được sau này. Còn với ung thư rời rạc đó là kết quả của sự đột biến soma ở cả 2 alen gen nguy cấp của cá thể.
Mặc dầu cơ sở di truyền của sinh khối u đã được thừa nhận, nhưng việc trị liệu thay thế gen lại không được cân nhắc trước tiên với tư cách như một chiến lược điều trị ung thư, bởi vì việc hiệu chỉnh một trong số các đa tổn thương dường như không thể làm cho khối u thoái lui. Các nghiên cứu về sự thay thế TSG trong các dòng tế bào và trên các động vật thí nghiệm những năm cuối 1980 và đầu 1990 tuy đã cho thấy những sai sót đáng ngờ, nhưng 3 khảo sát lâm sàng về thay thế gen p53 trong ung thư kết tràng và ung thư phổi vẫn được khởi đầu vào năm 1994. Đến cuối 2002, 25 quy trình thay thế TSG đã được phê chuẩn bao gồm 7 khảo sát pha II và 2 khảo sát pha IIIII. Những kết quả của các khảo sát lâm sàng đã nhanh chóng đưa ra bằng chứng của các nguyên lý bằng việc chứng minh có sự chuyển giao TSG tới các tế bào khối u, sự biểu hiện gen trong các tế bào đích, hiệu ứng gây độc tối thiểu và những bằng chứng sau cuối về thoái lui hay ổn định của các khối u đã được xử lý. Những khảo sát sau này đã chứng minh rằng việc thay thế TSG cũng hoàn lại cho các khối u sự nhạy cảm hơn đối với các hiệu ứng trị liệu DNA (bị hủy hoại), mở rộng ứng dụng việc thay thế TSG với các quy trình có sự kết hợp giữa trị liệu thay thế TSG với hóa trị liệu và xạ trị.
Nhờ những kết quả đầy hứa hẹn này trong lâm sàng mà đã đẩy nhanh tiến độ của các công trình thực nghiệm và kết quả của các nghiên cứu ở phòng thí nghiệm đã tạo điều kiện để đưa ra giả định mới là sự hủy hoại TSG xảy ra tương đối sớm trong diễn tiến của các tổn thương di truyền dẫn tới ung thư. Sự hủy hoại của TSG ứng viên phát hiện được ở biểu mô phổi bình thường và trong các tổn thương tiền u đã chứng minh rằng các TSG ngoài việc đem lại lợi ích trị liệu đối với các bệnh nhân ung thư nó còn có giá trị trong việc phòng ngừa và phát hiện sớm ung thư.
TÍNH CHẤT SINH HỌC VÀ HÓA SINH CỦA TSG
Người gác cổng (gatekeeper) và người quản gia (carekeeper)
Theo như bảng thống kê thì rõ ràng là chức năng chủ yếu của TSG là người gác cổng (gatekeeper) cho tế bào còn một số khác lại giống như người quản gia (caretaker) hơn (xem Bảng 3.1). Một số TSG như p53 lại biểu hiện cả chức năng người gác cổng cũng như là người quản gia. Nói chung, các TSG gác cổng sẽ điều hòa trực tiếp chức năng tế bào liên quan tới tăng trưởng, biệt hóa và chết của tế bào; các gen quản gia thì kiểm soát các quá trình tế bào liên quan tới việc sửa chữa các gen hư hỏng và duy trì sự toàn vẹn của hệ gen.
Các đột biến ở gen gác cổng sẽ trực tiếp dẫn đến ung thư, cho phép sự tăng sinh không kiểm soát và làm gián đoạn cảm ứng apoptosis; sự bất hoạt của các gen quản gia sẽ dẫn tới sự mất ổn định di truyền, kết quả là gây đột biến ở các gen khác kể cả các TSG gác cổng. Các ví dụ về gen gác cổng là p53, p73, PTEN, Fhit (Fragile histidine triad bộ ba histtidine dễ gẫy), Rb ( retinoblastoma) (u nguyên võng mạc), Von Lindau (bệnh u mạch võng mạc di truyền có đặc điểm kết hợp với u mạch tiểu não bẩm sinh; cũng có thể có các tổn thương tương tự ở tủy sống và các u nang ở tụy, thận và cácònội tạng khác. Triệu chứng thần kinh bao gồm: co giật, chậm phát triển trí tuệ), APC (adenomatous polyposis) và NF1 (neurofibromatous type 1) (u xơ thần kinh type 1). Các gen quản gia bao gồm ATM (ataxia telangiectasia mutated) (đột biến mất điều hòa giãn mao mạch), BRCA1, BRCA2, ATR (ATM and Rad3 related) và các gen sửa chữa những bất hợp lý. Tóm tắt chức năng của một số TSG hay biến đổi nhất trong ung thư và các bệnh liên quan với chúng được trình bày ở Bảng 1.
Những TSG có thể cảm ứng được apoptosis hoặc kiềm chế sự tăng trưởng khối u thường nhất là các gen gác cổng, đó là những đích hấp dẫn nhất đối với việc phát triển các chiến lược thay thế gen. Một số TSG rõ ràng gây cảm ứng apoptosis hay làm dừng chu kỳ tế bào khi được đưa vào các tế bào khối u như APC, RB, p16, p21, E2F1, PTEN, Fhit, p73 và TSG được lưu ý nhất hiện nay là p53.
Các TSG, con đường làm ngừng sự tăng trưởng và chết theo chương trình của tế bào
Vô số chức năng quan trọng của các TSG trong tế bào bình thường chưa được đề cập một cách chi tiết trong chương này. Tuy nhiên, có thể tóm tắt vai trò của p53 trong các con đường (pathway) tế bào sẽ giúp chúng ta hiểu rõ hơn về những lý do phía sau của việc trị liệu thay thế gen p53 cũng như thay thế hợp lý của các TSG khác.
Sự biểu hiện của một số sản phẩm gen bao gồm các yếu tố tăng trưởng, các gen gây ung thư, các cyclin và kinase phụ thuộc cyclin (cyclin-dependent kinase CDK) hướng tế bào (TB) tới tăng sinh. Sự biểu hiện của các TSG và các chất ức chế khác của CDK sẽ cảm ứng chu kỳ TB ngừng lại khi thích hợp, duy trì số lượng TB ở mức thích hợp đối với một mô cá biệt. Sự tăng sinh TB của động vật có vú hầu hết được điều hòa bởi 2 con đường liên hệ với nhau, đó là con đường Rb và con đường p53, cả 2 con đường này đều được điều hòa ở mức độ protein bởi các gen ung thư (oncogene) và cả các TSG khác. Nhìn chung, protein Rb (sản phẩm TSG của u nguyên bào võng mạc) điều hòa sự duy trì, giải phóng pha G1 còn protein p53 điều hành stress của TB và hủy hoại DNA hoặc gây ảnh hưởng tới sự ngừng tăng trưởng và sửa chữa hoặc là cảm ứng apoptosis. Khi hủy hoại một hoặc nhiều gen trong các con đường này có thể hướng TB tới việc sử dụng một con đường khác cho sự điều hòa tăng sinh, nếu không duy trì được con đường này nữa thì bắt đầu tăng sinh ngoài tầm kiểm soát.
Khi một TB bị stress do khởi động gen ung thư, suy giảm oxy mô hay hủy hoại DNA thì đó là nhiệm vụ của người bảo vệ hệ gen (guardian of the genome) p53, xác định xem TB sẽ nhận tín hiệu để tạm nghỉ ở giai đoạn G1 của chu kỳ TB hay sẽ nhận tín hiệu sửa chữa hoặc là sẽ tự hủy hoại mình thông qua apoptosis. Apoptosis giữ vai trò chính trong rất nhiều cơ chế của TB bình thường từ khi sinh phôi cho tới hủy hoại DNA gây bởi các đột biến ngẫu nhiên, bức xạ ion hóa và các chất hóa học gây hủy hoại DNA và cũng liên quan tới cơ chế chủ chốt trong chết theo chương trình của TB khi trị liệu ung thư (gây hủy hoại DNA) như hóa trị và xạ trị.
Sự cân bằng được duy trì một cách chính xác giữa 2 dạng tín hiệu nhận được bởi một TB ở một thời điểm bất kỳ - tiền apoptosis (proapoptosis) và tiền sống sót (prosurvival antiapoptotic) sẽ xác định apoptosis có bị cảm ứng hay không. Mặc dầu những tín hiệu này được xác định là do hoạt tính của protein p53, nhưng biểu hiện của nhiều gen sẽ nảy ra các tín hiệu nguy cấp mà các tín hiệu này lại được điều hòa bởi trạng thái hoạt hóa của p53, tạo nên vòng feedback phức tạp. P53 thực hiện nhiệm vụ giữ nhà (house keeping) bằng cách nhắm vào các gen tiền sống sót (prosurvival) hoặc kháng apoptosis (antiapoptotic), kể cả các proto -oncogene như bcl-2, bcl-X2, bcl-w và CED9 , cũng như các gen proapoptosis như bax, bad, và bid. Bản sao của mỗi gen này có thể tương tác với nhau tạo nên các dimer dị loại (heterodimer) và tỷ lệ tương đối giữa proapoptosis với các protein prosurvival trong các heterodimer sẽ xác định TB đang sống hay sẽ hướng tới apoptosis.
Con đường p53 được điều hòa bởi mức protein của các TSG khác và bởi một số gen ung thư (oncogene). Chẳng hạn như mdm2 (sản phẩm của proto -oncogene mdm2) gắn bình thường với N - terminal transactivating domain của p53, tiền ức chế hoạt hóa p53, dẫn tới phân rã nhanh của nó. Trong sự kiện stress gen độc tính kết quả là làm hủy hoại DNA do sự phosphoryl hóa serin trên p53, làm yếu liên kết với mdm2 và làm mất ổn định tương tác p53 / mdm2. Kết quả là làm tăng hoạt tính liên kết DNA trong p53 dẫn đến chuỗi tín hiệu điều chỉnh xuống gây nên việc đóng hoặc mở các gen khác.
Bảng 3.1. Các TSG gây gián đoạn thường gặp trong ung thư: chức năng bình thường và các bệnh liên quan tới mất chức năng.
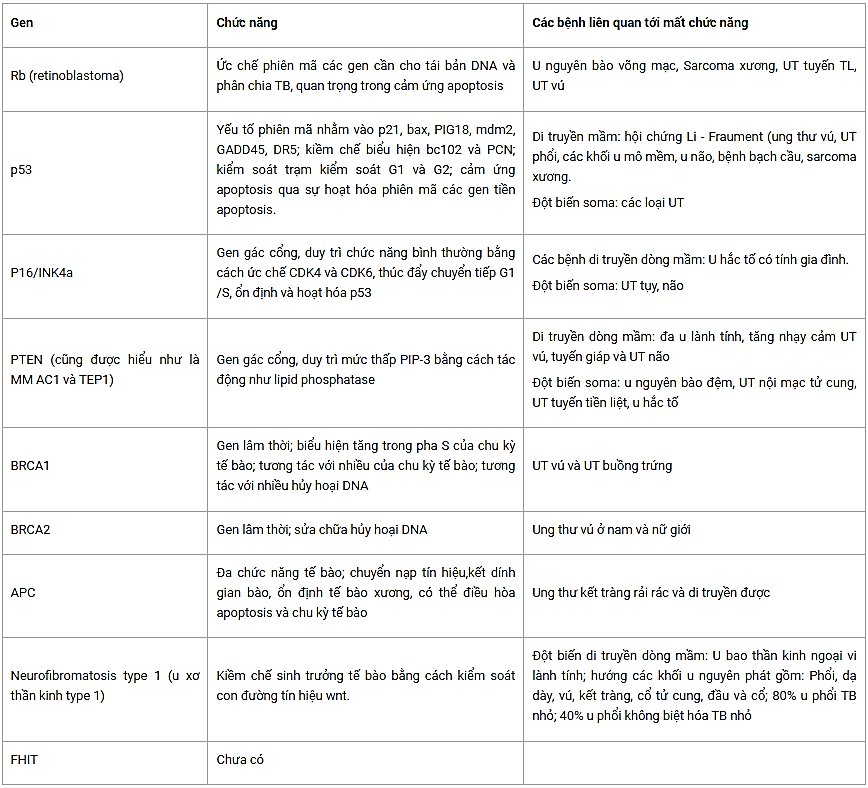
UT: Ung thư
Ở các TB bình thường, mdm2 bị ức chế bởi proto -oncogene làm cảm ứng biểu hiện pl19ARF - một TSG được mã hóa bởi cùng một locus gen như p16INK4a, nhưng lại đọc ở khung đọc luân phiên (alteRNAtive reading frame).
Vì p53 hoạt hóa có bán phần sống rất ngắn (khoảng 20 phút) nên việc gắn của mdm2 phải được kiểm soát để đảm bảo chắc chắn rằng p53 hoạt hóa phải ở mức rất thấp trong TB bình thường, vì thế đòi hỏi phải duy trì cân bằng giữa tăng sinh và ngừng chu kỳ TB hoặc apoptosis. Cả Rb và p53 đều bị biến đổi trong ung thư, vì thế một số TSG khác và proto -oncogene đều có liên quan tới các con đường này. Ví dụ như, khi loại bỏ TSG pl19ARF sẽ dẫn đến tăng mức mdm2 và tiếp theo là bất hoạt p53 dẫn đến tiến triển không thích hợp xuyên qua chu kỳ tế bào.
THAY THẾ TSG TRONG UNG THƯ
Thành công của các chiến lược thay thế gen đối với ung thư trước hết phải kể đến là việc chuyển thành công các gen trị liệu vào trong các mô đích dự định rồi sau đó biểu hiện được gen ở các mức độ thích hợp với độc tính thấp có thể chấp nhận được. Chỉ khi nào các đòi hỏi này đã được thỏa mãn thì mới định lượng được mức độ thoái lui của khối u và mới thỏa mãn được các ích lợi trị liệu. Một số TSG được đề cập ở đây đã tỏ rõ tiềm năng ứng dụng trong các thí nghiệm tiền lâm sàng tại phòng thí nghiệm và các nghiên cứu trên động vật và một số có thể được ứng dụng trong các thử nghiệm lâm sàng. Bởi vì hầu hết các thử nghiệm lâm sàng cho tới nay đều liên quan tới việc thay thế các TSG dễ bị biến đổi nhất trong ung thư như p53 chẳng hạn, vì vậy chúng ta sẽ bàn luận về các nghiên cứu tiền lâm sàng và sau đó là các thử nghiệm lâm sàng hướng vào gen này. Những tiến bộ đạt được khi thay thế gen p53 cũng tạo điều kiện cho chúng ta hiểu rõ hơn về các ứng dụng tiềm năng của các TSG khác đối với trị liệu ung thư.
Trị liệu thay thế TSG: trường hợp p53
Lý do cơ bản:
Mất chức năng p53 quan sát thấy ở mức trên 50% với tất cả các u ác tính, đó là tổn thương di truyền thông thường nhất trong ung thư. Nếu sau đó ta cài một bản sao gen hoang dã thì cũng đủ để hoàn trả sự cân bằng bình thường giữa tăng sinh và chết theo chương trình của tế bào đối với một TB khối u. Hơn nữa, vai trò đặc biệt của p53 trong việc cảm ứng sự chết theo chương trình của TB chỉ rõ rằng việc thay thế gen này có thể làm hoàn trả hoặc tăng cường tính nhạy cảm đối với các tác nhân trị liệu như hóa trị, xạ trị - tùy thuộc vào sự hủy hoại DNA và apoptosis phụ thuộc p53. Nó được coi như một cơ chế phá hủy các TB khối u.
Xem tiếp: Thay thế các gen kiềm chế khối u trong ung thư (P2)
Bệnh viện Nguyễn Tri Phương - Đa khoa Hạng I Thành phố Hồ Chí Minh
BÀI VIẾT KHÁC


