

️ Bệnh thalassemia thể trung gian và HbE (P1)
ĐỊNH NGHĨA
Kiểu hình lâm sàng của thalassemia thể trung gian nằm giữa thể nhẹ (dị hợp tử) và thể nặng (đồng hợp tử), mặc dù nhiều đặc điểm lâm sàng có thể trùng lắp giữa ba thể trên. Năm 1955, lần đầu tiên Rietti-GreppiMicheli dùng từ thalassemia thể trung gian để mô tả những bệnh nhân - về mặt huyết học - chưa đủ để gọi là thể nặng nhưng cũng không thể gọi là thể nhẹ.
Thalassemia thể trung gian có đặc điểm lâm sàng đa dạng. Bệnh nhân thể nhẹ hoàn toàn không có triệu chứng cho đến lớn; hoặc chỉ biểu hiện thiếu máu nhẹ với mức Hb từ 7-10 g/dL. Những bệnh nhân này chỉ cần truyền máu vài lần. Bệnh nhân thể nặng hơn thường có biểu hiện lâm sàng từ 2-6 tuổi, mặc dù bệnh nhân có thể sống mà không cần truyền máu thường xuyên, nhưng sự tăng trưởng và phát triển có thể chậm. Do đặc điểm lâm sàng đa dạng của thalassemia thể trung gian nên việc điều trị cũng tùy trường hợp cụ thể. Dù có nhiều phương pháp điều trị để chọn lựa nhưng sự thiếu hụt phác đồ hướng dẫn cụ thể được xem là một thách thức đáng kể trong lâm sàng (Taher, 2006; Camaschella và Cappellini, 1995).
CƠ CHẾ SINH BỆNH CỦA THALASSEMIA THỂ TRUNG GIAN
Sinh bệnh học của thalassemia dựa trên sự mất cân bằng tổng hợp chuỗi globin. Trong β-thalassemia thể trung gian, sự mất cân bằng thấy rõ hơn so với thể nhẹ nhưng lại ít hơn so với thể nặng. Hầu hết bệnh nhân thalassemia thể trung gian là β-thalassemia thể đồng hợp tử hoặc phức hợp dị hợp tử, nghĩa là cả hai locus của gen chuỗi β-globin đều bị ảnh hưởng. Biểu hiện lâm sàng của thalassemia thể trung gian nhẹ so với thalassemia thể nặng chủ yếu là do ba cơ chế sau:
- Di truyền của đột biến β+ nhẹ;
- Hiện diện polymorphism của enzyme Xmn-I tại vùng hoạt hóa Gγ, liên quan đến việc gia tăng HbF;
- Đồng di truyền của α-thalassemia trên locus của gen chuỗi β-globin.
Thalassemia thể trung gian cũng có thể là hậu quả của sự gia tăng sản xuất chuỗi α-globin, do sự nhân ba của gen tạo chuỗi β dị hợp tử hoặc do sự tương tác của β- và δβ- thalassemia (Taher, 2006).
Phân tích các kiểu gen của bệnh nhân thalassemia thể trung gian là rất quan trọng vì chẩn đoán sớm thể nhẹ có thể tránh truyền máu không cần thiết.
Dự đoán kiểu hình từ kiểu gen của bệnh nhân thalassemia thể trung gian vẫn còn khó khăn, do sự tương tác của các yếu tố di truyền và môi trường. Những yếu tố gây biến đổi di truyền cấp một là vô số các gen a-len trên locus của chuỗi β- có thể gây giảm một phần hoặc hoàn toàn tổng hợp chuỗi β-. Những yếu tố gây biến đổi di truyền cấp hai là việc ảnh hưởng trực tiếp lên số lượng của các chuỗi α- dư thừa (di truyền của gen chuỗi α- hay γ- bất thường). Những yếu tố gây biến đổi di truyền cấp ba thì rất đa dạng - xảy ra trên các locus liên quan đến sự chuyển hóa xương, sắt và bilirubin - có thể ảnh hưởng đến biểu hiện lâm sàng. Yếu tố môi trường có liên quan bao gồm điều kiện xã hội, dinh dưỡng và chăm sóc y tế sẵn có (Taher, Ismaeel và Cappellini, 2006).
Chẩn đoán phân biệt
Chẩn đoán phân biệt giữa thalassemia thể nặng và thể trung gian là cần thiết để chọn phương pháp điều trị phù hợp cho từng trường hợp. Dự đoán chính xác một kiểu hình nhẹ có thể tránh việc truyền máu không cần thiết và các biến chứng của nó. Trong khi đó, chẩn đoán thalassemia thể nặng đúng thời điểm cho phép bắt đầu truyền máu sớm, do đó có thể ngăn cản hoặc làm chậm diễn tiến cường lách và giảm nguy cơ tạo kháng thể kháng hồng cầu. Nhưng không may, việc xác định chính xác hai kiểu hình này tại thời điểm mới khởi phát bệnh còn rất khó khăn. Tuy nhiên, phân tích chi tiết về lâm sàng, huyết học, gen và dữ liệu phân tử có thể cho phép chọn lựa phương pháp điều trị hợp lý (Taher, 2006; Wainscoat, 1987; Weatherall, 2001). (xem Bảng 1 để thấy những điểm khác nhau chính giữa thalassemia thể trung gian và thể nặng).
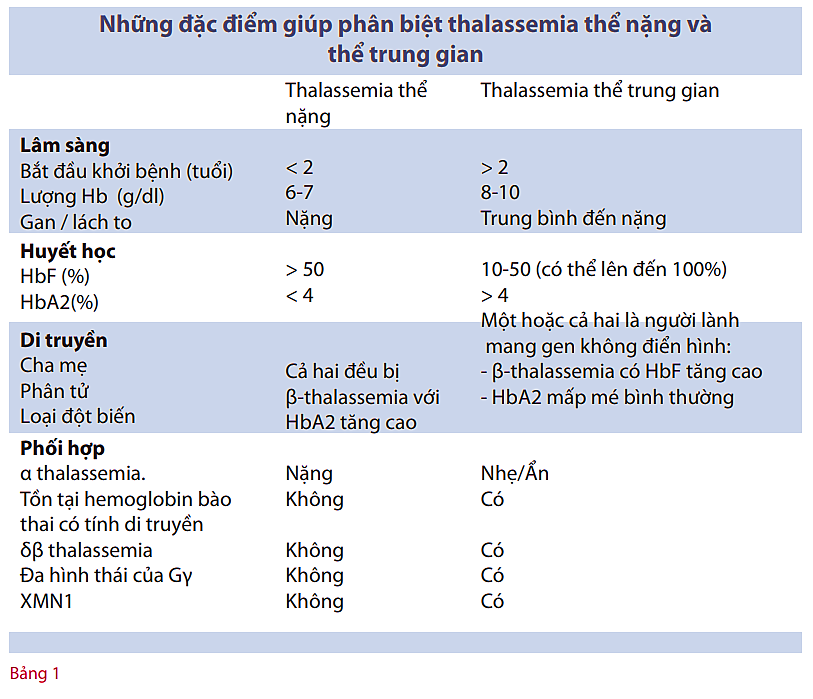
Sinh lý bệnh của thalassemia thể trung gian
Ba yếu tố chính gây ra các triệu chứng lâm sàng của thalassemia thể trung gian: sự tạo hồng cầu không hiệu quả, thiếu máu mạn tính và ứ sắt. Mức độ nặng của triệu chứng phụ thuộc chính vào mức độ khiếm khuyết phân tử. Các chuỗi a- có tính rất không ổn định và lắng đọng trong các tế bào tiền thân hồng cầu ở tủy xương gây tổn thương màng tế bào và chết tế bào (tức là sự tạo hồng cầu không hiệu quả).
Sự phì đại của phần tủy tạo hồng cầu trong tủy xương và phần ngoài tủy dẫn đến biến dạng điển hình của xương sọ và mặt, mỏng vỏ xương và gãy xương bệnh lý ở các xương dài. Mức độ tạo hồng cầu không hiệu quả là yếu tố chính gây ra thiếu máu, trong khi đó, sự tán huyết của hồng cầu trưởng thành ở máu ngoại vi và giảm tổng hợp hemoglobin là chỉ là yếu tố phụ.
CÁC BIẾN CHỨNG VÀ ĐIỀU TRỊ BỆNH NHÂN THALASSEMIA THỂ TRUNG GIAN
Ngoài các triệu chứng đã được mô tả của thalassemia thể trung gian, các triệu chứng có thể biểu hiện nhẹ hơn hoặc nặng hơn ở thalassemia các thể khác. Kinh nghiệm cho thấy, bệnh nhân thalassemia thể trung gian có một số biến chứng đặc biệt hiếm gặp ở thalassemia thể nặng. Hình 1 mô tả các điểm nổi bật về biến chứng của bệnh thalassemia không được điều trị (Taher, Ismaeel và Cappellini, 2006; Cappellini, Cerino và cs., 2001).
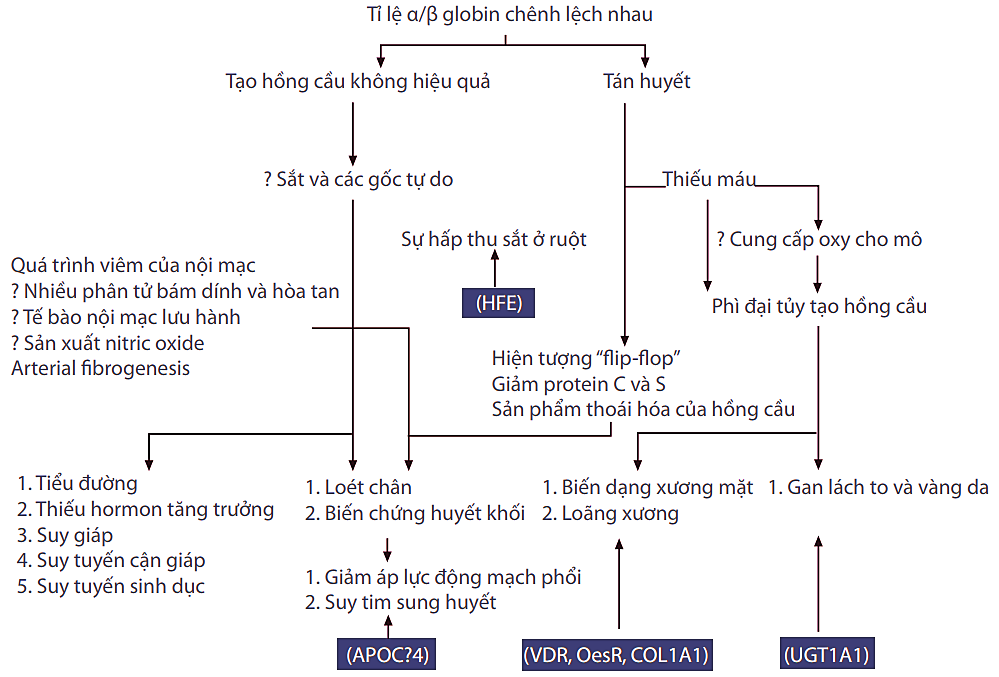
Hình 1: Chuỗi sinh bệnh học của bệnh thalassemia không được điều trị và các biểu hiện lâm sàng tương ứng
Lách to và phẫu thuật cắt lách
Phẫu thuật cắt lách ngày nay không còn phổ biến và chủ yếu được thực hiện ở giai đoạn sau của bệnh. Chỉ định cắt lách trong thalassemia thể trung gian khi lách quá to và giảm nồng độ Hb trung bình trong máu đồng thời không kèm theo các yếu tố tạm thời khác như nhiễm khuẩn (Taher, Ismaeel và Cappellini, 2006; Cappellini, Cerino và cs., 2001; Borgna -Pignatti, Rigon, Merlo và cs., 2003; Galanello, Piras, Barella và cs., 2001). Đối với các loại phẫu thuật, phương pháp nội soi là an toàn, khả thi và được ưu tiên hơn phẫu thuật hở. Đây là phương pháp xâm lấn tối thiểu và được ưu tiên lựa chọn trong điều trị bệnh nhân β-thalassemia có chỉ định cắt lách. Khi tiến hành cắt lách, phẫu thuật viên cần kiểm tra túi mật để tìm sỏi và thực hiện cắt túi mật ngay khi tìm thấy sỏi túi mật (Leandros và cs., 2006).
Sỏi mật và phẫu thuật cắt túi mật
Sỏi mật thường gặp trong thalassemia thể trung gian nhiều hơn so với thể nặng, là do sự tạo hồng cầu không hiệu quả và tán huyết ở máu ngoại biên. Tương tự như cắt lách qua nội soi, nội soi cắt túi mật tiện lợi và khả thi hơn phẫu thuật hở (Taher, Ismaeel và Cappellini, 2006; Cappellini, Cerino và cs., 2001; Borgna-Pignatti, Rigon, Merlo và cs., 2003; Galanello, Piras, Barella và cs., 2001; Leandros và cs., 2006).
Sự tạo máu ngoài tủy
Tạo máu ngoài tủy là cơ chế bù trừ do tủy xương hoạt động quá mức để đáp ứng tình trạng thiếu máu mạn trong thalassemia thể trung gian. Từ đó, hình thành các khối mô tạo hồng cầu mà chủ yếu là ở lách, gan, các hạch bạch huyết, ngực và cột sống. Các khối này có thể được phát hiện bằng chụp cộng hưởng từ (MRI) và có thể gây ra các vấn đề về thần kinh như chèn ép tủy sống, liệt hai chi dưới, khối u trong ngực.
Trong trường hợp bị chèn ép cột sống, nhận biết các dấu hiệu lâm sàng là rất quan trọng để chẩn đoán sớm và phòng ngừa các biến chứng thần kinh không hồi phục. MRI là phương pháp chẩn đoán hình ảnh được lựa chọn để chẩn đoán các khối u tạo máu ngoài tủy đồng thời có thể thấy hình ảnh phì đại của phần tủy sống liên quan.
Việc điều trị bao gồm truyền máu, xạ trị và hydroxyurea (Taher, Ismaeel và Cappellini, 2006; Chehal, Aoun, Koussa và cs., 2003; Castelli, Graziadei, Karimi và Cappellini, 2004; Saxon, Rees, Olivieri, 1998). Tăng cường truyền máu là phương pháp điều trị đầy hứa hẹn, nhằm mục tiêu đưa Hb lên mức cao hơn, bao gồm việc truyền máu nhiều lần mỗi vài tuần để bù đắp cho nhu cầu tạo hồng cầu.
Sỏi thận
Bệnh nhân thalassemia thể trung gian thường dễ bị sỏi thận do hậu quả của việc tạo hồng cầu không hiệu quả và tán huyết ngoại biên. Sỏi thận có thể dẫn đến thận ứ nước và suy thận. Nguyên nhân là do sỏi phì đại làm tắc nghẽn các ống thận và thậm chí cả đài thận. Thận thường phì đại trong bệnh nhân thalassemia do sự tạo máu ngoài tủy.
Loét chân
Loét chân thường gặp ở bệnh nhân lớn tuổi hơn là trẻ nhỏ trong thalassemia thể trung gian. Cơ chế không rõ nhưng một khi loét đã bắt đầu tiến triển thì rất đau đớn và khó chữa. Tuy nhiên truyền máu thường xuyên có thể làm thuyên giảm phần nào trong những trường hợp bệnh kéo dài. Việc bổ sung kẽm có thể giúp đẩy nhanh tiến trình chữa lành vết loét. Hydroxyurea cũng có nhiều lợi ích, có thể dùng một mình hoặc kết hợp với erythropoietin hoặc yếu tố tăng trưởng có nguồn gốc từ tiểu cầu. Ngoài ra, sử dụng buồng oxy để điều trị có thể làm cải thiện triệu chứng vì thiếu oxy mô có thể là nguyên nhân chính của các vết loét (Taher, Ismaeel và Cappellini, 2006; Gimmon, Wexler và Rachmilewitz, 1982).
Huyết khối
Bệnh nhân thalassemia thể trung gian có khuynh hướng dễ bị huyết khối hơn thể nặng. Huyết khối chủ yếu hình thành ở hệ thống tĩnh mạch, bao gồm huyết khối tĩnh mạch sâu (40%), huyết khối tĩnh mạch cửa (19%), nhồi máu não (9%), nghẽn mạch phổi (12%) và các tĩnh mạch khác (20%). Hơn nữa, những bệnh nhân đã cắt lách có nguy cơ tắc mạch cao hơn so với những bệnh nhân không cắt lách (Cappellini, Robbiolo, Bottasso và cs., 2000). (Xem Hình 2 để biết thêm cơ chế tạo huyết khối trong thalassemia thể trung gian).
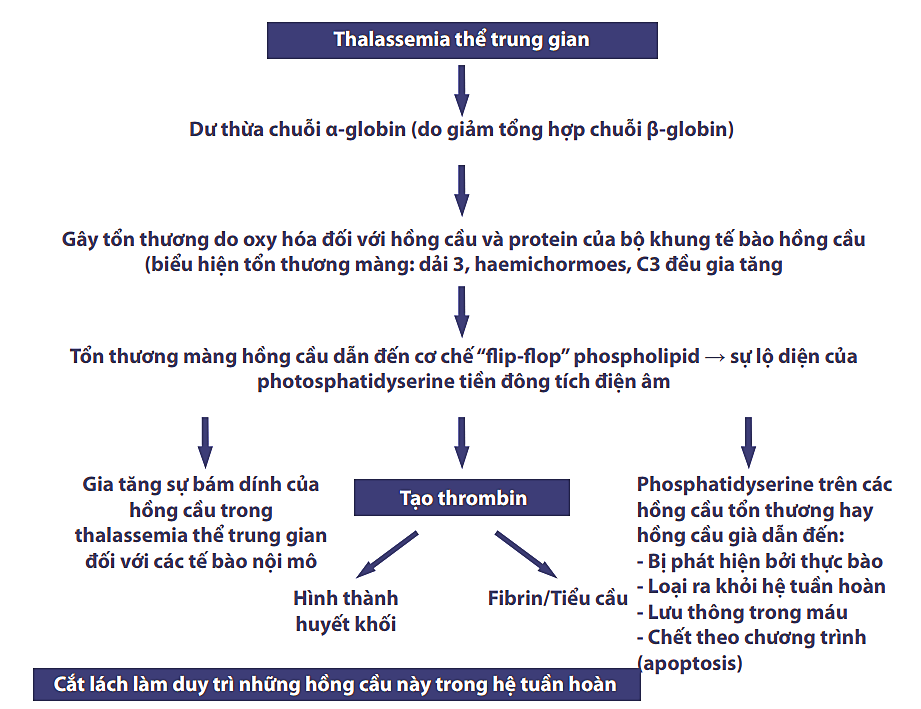
Hình 2: Cơ chế huyết khối trong thalassemia thể trung gian
Điều trị bệnh nhân bị huyết khối gồm hai nhánh chính: phòng ngừa và điều trị. Phòng ngừa bằng cách dùng thuốc chống đông phù hợp trước phẫu thuật hoặc những thủ thuật có nguy cơ cao. Điều trị đòi hỏi phải sử dụng đầy đủ thuốc kháng đông theo khuyến cáo dành cho bệnh nhân dễ bị tăng đông. Nhận thức rõ vấn đề huyết khối là rất quan trọng vì sự nghẽn mạch do huyết khối đóng vai trò quan trọng trong tăng áp động mạch phổi và suy tim phải (Taher, Ismaeel và Cappellini, 2006; Taher, Ismaeel, Mehio, Bignamini và cs., 2006; Eldor, Rachmilewitz, 2002; Cappellini, Robbiolo, Bottasso và cs., 2000; Taher, Abou-Mourad, Abchee và cs., 2002).
Tăng áp động mạch phổi và suy tim ứ huyết
Tăng áp động mạch phổi rất thường gặp ở bệnh nhân thalassemia thể trung gian. Một nghiên cứu trên 110 bệnh nhân thalassemia thể trung gian (60,9% không cần truyền máu hoặc truyền rất ít) thấy 59,1% số bệnh nhân có dấu hiệu tăng áp động mạch phổi và được cho là nguyên nhân chính của suy tim sung huyết trên các bệnh nhân trong mẫu nghiên cứu (Aessopos, Farmakis, Karagiorga và cs., 2001). Cơ chế chính gây tăng áp động mạch phổi trong thalassemia thể trung gian vẫn còn chưa rõ ràng.
Vì thiếu máu và ứ sắt không thường gặp ở những bệnh nhân thalassemia thể nặng được truyền máu và thải sắt tốt, nên hai tình trạng trên có thể là nguồn gốc sinh lý bệnh của tăng áp động mạch phổi. Vì vậy những bệnh nhân thalassemia thể trung gian khi có dấu hiệu sớm của tăng áp động mạch phổi cần được phân nhóm để được chỉ định truyền máu thường xuyên và thải sắt. Sildenafil cũng đã được sử dụng thành công để điều trị tăng áp động mạch phổi mặc dù nghiên cứu được tiến hành trên lượng lớn bệnh nhân nhưng thiếu những bệnh nhân thalassemia thể trung gian (Taher, Ismaeel và Cappellini, 2006; Aessopos, Farmakis, Karagiorga và cs., 2001; Aessopos, Farmakis, Deftereos và cs., 2005).
Viêm gan
Viêm gan do virus (B và C) ít gặp ở bệnh nhân thalassemia thể trung gian hơn thể nặng vì ít phải truyền máu thường xuyên hơn. Men gan tăng bất thường (AST và ALT) thường thấy ở bệnh nhân thalassemia thể trung gian, chủ yếu là vì tế bào gan bị tổn thương do lắng đọng chất sắt. Men gan thường trở về bình thường khi điều trị thải sắt đầy đủ (Taher, Ismaeel và Cappellini, 2006; Cappellini, Cerino, Marelli và Fiorelli, 2001).
Chức năng nội tiết
Suy sinh dục, suy giáp và tiểu đường dường như rất hiếm gặp trong thalassemia thể trung gian. Mặc dù bệnh nhân thalassemia thể trung gian thường dậy thì muộn, họ vẫn phát triển sinh dục bình thường và có khả năng thụ tinh. Một vài trường hợp có thể suy giáp khi về già (Taher, Ismaeel và Cappellini, 2006; Cappellini, Cerino, Marelli và Fiorelli, 2001).
Vấn đề mang thai ở bệnh nhân thalassemia thể trung gian
Bệnh nhân nữ thalassemia thể trung gian có thể mang thai tự nhiên thành công, mặc dù biến chứng trong thai kỳ có thể xảy ra. Tình trạng thiếu máu mạn trong thalassemia thể trung gian có thể làm tăng tần suất xảy thai tự nhiên, sinh non và thai chậm phát triển trong tử cung, trong khi đó, các biến chứng về nội tiết thường do lắng đọng hemosiderin. Thiếu hụt acid folic thường gặp trong thalassemia thể trung gian do sự hấp thụ kém, chế độ ăn thiếu cung cấp hoặc đáng kể nhất là do tăng nhu cầu acid folic để đáp ứng với tình trạng tủy xương tăng hoạt động. Trong thời gian mang thai, phụ nữ bị thalassemia thể trung gian phải được uống bổ sung acid folic (khoảng 1 mg/ngày), và nên được theo dõi cẩn thận để truyền máu khi cần thiết, tránh tình trạng rối loạn huyết động học do thiếu máu (Taher, Ismaeel và Cappellini, 2006; Nassar, Rechdan, Usta và Taher, 2006).
Ứ sắt
Cũng giống như thalassemia thể nặng, bệnh nhân thalassemia thể trung gian dễ bị biến chứng ứ sắt. Tuy nhiên, cơ chế ứ sắt trong thalassemia thể trung gian là do tăng hấp thu ở ruột chứ không phải do truyền máu. Hậu quả của việc ứ sắt có thể dẫn đến một số biến chứng nghiêm trọng bao gồm suy tim, bất thường nội tiết như tiểu đường và suy tuyến sinh dục (Taher, Ismaeel và Cappellini, 2006; Weatherall, 2001).
Khởi đầu của việc thải sắt phụ thuộc lượng sắt dư, tỉ lệ tích tụ sắt và thời gian phát hiện ứ sắt. Sự tăng nồng độ sắt trong gan (LIC) đã được ghi nhận cùng với sự tăng nhẹ ferritin trong huyết thanh (Fiorelli, Fargion, Piperno và cs., 1990). Do đó, để trực tiếp đánh giá LIC nên làm sinh thiết gan hoặc MRI. Liệu pháp thải sắt nên được bắt đầu nếu LIC ≥ 7 mg/g trọng lượng khô của gan (Taher, Ismaeel và Cappellini, 2006).
Loãng xương
Trong thalassemia thể trung gian, có một tỉ lệ lớn bệnh nhân bị loãng xương cột sống và xương chậu ở cả hai giới. Độ nặng của loãng xương tăng theo tuổi và có thể có ở những người trẻ tuổi nhưng mật độ khoáng trong xương cột sống thấp hơn rất nhiều so với những người cùng tuổi. Điều trị bao gồm bổ sung bisphosphonates, calci và theo dõi độ đậm đặc của xương (Origa, Fiumana và cs., 2005).
Pseudoxanthoma elasticum (PXE)
PXE là một rối loạn mô liên kết di truyền hiếm gặp, đặc trưng bởi sự thoái hóa toàn bộ các sợi đàn hồi với biểu hiện kiểu hình đa dạng. Đặc điểm lâm sàng biểu hiện chủ yếu ở da, mắt và mạch máu. Giải phẫu bệnh ở da cho thấy da bị sưng phù, các sợi đàn hồi gãy làm nhiều mảnh và kết cụm bất thường ở lớp lưới trung và sâu của hạ bì, đồng thời lắng đọng calci thứ phát. Hiện tượng này đã được mô tả trong bệnh thalassemia.
Xem tiếp: Bệnh thalassemia thể trung gian và HbE (P2)
Bệnh viện Nguyễn Tri Phương - Đa khoa Hạng I Thành phố Hồ Chí Minh
BÀI VIẾT KHÁC


