

️ Bệnh thalassemia thể trung gian và HbE (P2)
ĐIỀU TRỊ BỆNH NHÂN THALASSEMIA THỂ TRUNG GIAN
Hiện nay có nhiều cách chọn lựa cho điều trị bệnh nhân thalassemia thể trung gian bao gồm cắt lách, truyền máu, điều hòa sản xuất Hb bào thai và ghép tủy xương (Taher, Ismaeel và Cappellini, 2006; Cappellini, Cerino, Marelli và Fiorelli, 2001).
Cắt lách
Cắt lách không còn là vấn đề chủ yếu trong điều trị bệnh. Tuy nhiên, những chỉ định cắt lách chính bao gồm chậm phát triển hoặc sức khỏe kém, giảm bạch cầu, giảm tiểu cầu, tăng nhu cầu truyền máu và lách to có triệu chứng.
Cắt lách trước 5 tuổi nguy cơ nhiễm trùng cao nên không được khuyến cáo.
Liệu pháp truyền máu và thải sắt
Mặc dù truyền máu hiện không phải là liệu pháp điều trị thường xuyên cho các bệnh nhân thalassemia thể trung gian nhưng nó vẫn có những lợi ích đáng kể. Quyết định bắt đầu điều trị nên dựa trên mức độ nghiêm trọng của triệu chứng thiếu máu, kể cả dấu hiệu chậm tăng trưởng và phát triển. Vì mức độ ứ sắt thay đổi trong thalassemia thể trung gian, nên việc đánh giá nồng độ sắt trong gan là việc nên làm trước khi bắt đầu liệu pháp truyền máu. Bệnh nhân thalassemia thể trung gian có thể sẽ tốt hơn nếu có một chế độ truyền riêng cho từng trường hợp bệnh nhân, khác với chế độ truyền thường xuyên trong thalassemia thể nặng, để ngăn ngừa việc lệ thuộc truyền máu. Phản ứng miễn dịch do kháng thể bất thường tương đối thường gặp trong thalassemia thể trung gian, mặc dù nguy cơ trên sẽ giảm nếu bắt đầu truyền máu trước 12 tháng tuổi (Pippard, Callender, Warner và Weatherall, 1979; Mourad, Hoffbrand, Sheikh-Taha và cs., 2003; Cappellini, 2001).
Truyền máu được chỉ định khi thấy những dấu hiệu sau:
- Trẻ không phát triển với những biểu hiện rõ của thiếu máu;
- Biểu hiện biến dạng xương;
- Tăng thiếu máu không do các nguyên nhân có thể tự hồi phục;
- Có biểu hiện lâm sàng tắc mạch;
- Xuất hiện các vết loét ở chân;
- Tăng áp động mạch phổi tiến triển;
- Chậm hoặc dậy thì trễ;
- Lách to tiến triển.
Điều hòa sự sản xuất hemoglobin bào thai
Tăng tổng hợp Hb bào thai có thể làm giảm thiếu máu và do đó cải thiện tình trạng lâm sàng của bệnh nhân thalassemia thể trung gian. Các chất như cytosine arabinoside và hydroxyurea có thể làm thay đổi kiểu tạo hồng cầu và tăng sự hoạt động của gen tạo chuỗi γ-. Erythopoietin tỏ ra có hiệu quả và có thể có tác dụng cộng thêm khi kết hợp với hydroxyurea. Butyrates là thử nghiệm điều trị chuyên sâu hơn, nhưng chưa được cấp phép và khó sử dụng. Nhiều phản hồi tốt được báo cáo, tuy nhiên, hầu hết bệnh nhân than phiền về khó khăn của việc uống thuốc và chích thuốc đường tĩnh mạch. Cần có những đánh giá lâm sàng chuyên sâu hơn nữa để làm rõ giá trị của hướng điều trị mới này (Taher, Ismaeel và Cappellini, 2006; Karimi, H. Darzi, M. Yavarian, 2005; Dettelbach và Aviado, 1985; Dixit, Chatterjee, Mishra và cs., 2005; Perrine, Ginder, Faller và cs., 1993; Cappellini, Graziadei, Ciceri và cs., 2000; Olivieri, Rees, Ginder và cs., 1997).
Ghép tủy xương
Ghép tủy xương là điều trị được thiết lập cho bệnh β-thalassemia. Mặc dù ghép tủy có thể điều trị hết bệnh nhưng mức độ của sự thành công còn phụ thuộc vào tình trạng sức khỏe và tuổi của bệnh nhân. Quyết định chọn bệnh nhân hội đủ điều kiện cho việc ghép cũng rất phức tạp và liên quan đến chất lượng cuộc sống, thời gian tiên lượng sống (thời gian sống mong đợi) của bệnh nhân được ghép tủy. Điều này đặc biệt liên quan đến bệnh nhân thalassemia thể trung gian, nhất là ở những bệnh nhân chỉ bị ảnh hưởng nhẹ. Ở bệnh nhân thalassemia thể trung gian ổn định không có triệu chứng và không cần truyền máu thì việc ghép tủy xương là không cần thiết.
Khuyến cáo về điều trị bệnh nhân thalassemia thể trung gian
Hai vấn đề lớn về điều trị bệnh nhân thalassemia thể trung gian:
- Hướng tiếp cận và điều trị các biến chứng ở bệnh nhân thalassemia thể trung gian là người lớn
- Ngăn chặn các biến chứng ở trẻ em.
Do đó, sự phân tầng về quản lý thalassemia thể trung gian giữa người lớn và trẻ em được thiết lập.
Kế hoạch điều trị cho các bệnh nhân thalassemia thể trung gian ở người lớn như sau:
- Mỗi bệnh nhân được xem xét riêng biệt và phân nhóm nguy cơ;
- Hydroxyurea được dùng như là cách tiếp cận khởi đầu thích hợp;
- Liệu pháp truyền máu kèm thải sắt với deferoxamine truyền dưới da và phối hợp với steroid để chống phản ứng miễn dịch do kháng thể bất thường là rất cần thiết;
- Dùng aspirin để phòng đột quỵ, dùng kháng đông kéo dài và sau cắt lách ở bệnh nhân có tiền căn huyết khối;
- Chụp MRI gan để đánh giá tình trạng ứ đọng sắt (hoặc sinh thiết gan nếu không thể làm MRI) là rất quan trọng để quyết định thải sắt trong tương lai
Không có một hướng dẫn rõ ràng cho việc quản lý thalassemia thể trung gian ở những người trẻ. Vì vậy, các tác giả khuyến cáo như sau:
- Đánh giá có cần phải cắt lách không và trì hoãn việc bắt đầu truyền máu trừ khi thực sự cần thiết, dựa vào những chỉ định được đề cập ở trên;
- Quyết định bắt đầu truyền máu sớm và liệu pháp thải sắt nếu bệnh nhân có bằng chứng tăng trưởng bất thường, học kém hoặc ảnh hưởng tâm lý thứ phát do biến dạng khuôn mặt;
- Thực hiện thường xuyên siêu âm tim, theo dõi biến chứng tim mạch và bắt đầu điều trị ngay khi bệnh khởi phát để ngăn ngừa sự tiến triển của bệnh;
- Theo dõi nồng độ sắt trong gan bằng MRI thường xuyên, hoặc sinh thiết gan;
- Khuyến cáo bệnh nhân không hút thuốc, bất động tư thế kéo dài, không nên dùng thuốc tránh thai đường uống hoặc vòng tránh thai.
Xem Bảng 2 về chỉ định truyền máu và cắt lách.
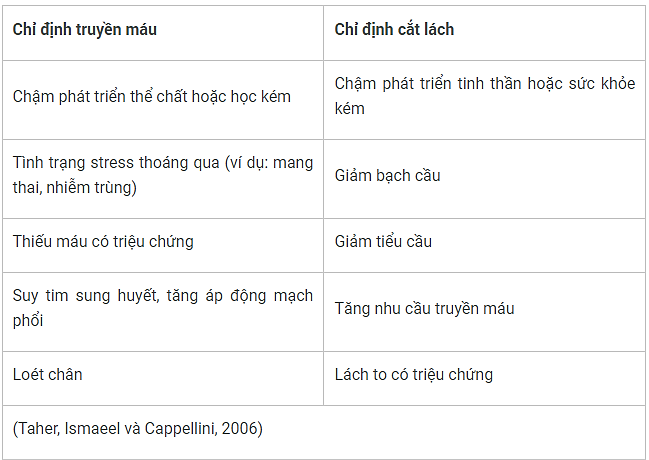
Bảng 2: Chỉ định truyền máu và cắt lách ở bệnh nhân thalassemia thể trung gian
β thalassemia/HbE
Hemoglobin E có đặc điểm lâm sàng của β-thalassemia mức độ nhẹ và thường gặp nhất ở Đông Nam Á, đặc biệt là phía đông Thái Lan và Lào. Sự kết hợp của HbE với β-thalassemia làm cho lâm sàng trở nên đa dạng, có thể thay đổi từ giống như thalassemia thể nặng đến dạng nhẹ của thalassemia thể trung gian (TIF, 2002; Premawardhena và cs., 2005).
Về mặt lâm sàng, β-thalassemia/HbE có thể được phân thành ba mức độ, mỗi mức độ có yêu cầu quản lý về mặt lâm sàng riêng.
β -thalassemia/ HbE thể nhẹ
β-thalassemia/HbE thể nhẹ không cần điều trị và hiếm khi biểu hiện các triệu chứng lâm sàng. Hemoglobin có thể cao tới 9-12 g /dl. Nên cẩn thận để không nhầm lẫn nhóm bệnh này với thiếu máu thiếu sắt hoặc người lành mang gen β-thalassemia. Có thể phân biệt bằng cách xem hình dạng hồng cầu, tình trạng sắt và điện di hemoglobin (TIF, 2002; và cs. Premawardhena, 2005).
β-thalassemia/HbE thể trung bình nặng
Nhóm này bao gồm phần lớn các bệnh nhân β-thalassemia/HbE. Hầu hết các bệnh nhân này có nồng độ hemoglobin ổn định ở mức 6-7 g/dl. Nhóm này có biểu hiện triệu chứng lâm sàng tương tự thalassemia thể trung gian và thường không cần phải truyền máu trừ khi bệnh nhân bị nhiễm trùng làm thúc đẩy thiếu máu. Các biến chứng khác như ứ sắt có thể xảy ra. Trong trường hợp đó nên bắt đầu thải sắt. Bệnh nhân trong nhóm này thường có tuổi thọ ngắn hơn, nhưng nếu được theo dõi và điều trị cẩn thận thì có thể có tiên lượng tốt hơn (TIF, 2002; Premawardhena và cs., 2005).
β thalassemia/HbE thể nặng
Bệnh nhân biểu hiện các triệu chứng lâm sàng của thalassemia thể nặng bao gồm chậm phát triển thể chất, biến dạng xương với những thay đổi trên xương mặt, thiếu máu, vàng da và gan lách to. Hb ở mức thấp khoảng 4-5 g/dl. Quản lý lâm sàng nhóm bệnh nhân này cũng giống như thalassemia thể nặng (TIF, 2002; Premawardhena và cs., 2005).
Biến chứng và việc quản lý β-thalassemia/HbE
Các biến chứng của bệnh nhân β-thalassemia/HbE phụ thuộc vào mức độ phân loại như đã nói ở trên. Xấu nhất là biến chứng xảy ra trong nhóm nặng với đặc điểm lâm sàng tương tự như β-thalassemia thể nặng. Biến chứng xảy ra bao gồm những vấn đề liên quan đến ứ sắt do việc lệ thuộc truyền máu.
Bệnh viện Nguyễn Tri Phương - Đa khoa Hạng I Thành phố Hồ Chí Minh
BÀI VIẾT KHÁC


