

️ Cơ sở di truyền và sinh lý bệnh Thalassemia
CÁC LOẠI HEMOGLOBIN
Oxy được vận chuyển từ phổi đến mô bởi một phân tử protein đặc biệt là hemoglobin (Hb) nằm trong hồng cầu. Mỗi hồng cầu chứa khoảng 300 triệu phân tử protein này, tương đương tổng cộng khoảng 30 pg/ hồng cầu. Mỗi phân tử Hb được thành lập bởi hai cặp tiểu đơn vị giống nhau là các chuỗi globin được đặt tên theo thứ tự chữ cái Hy Lạp và thuộc vào hai nhóm: nhóm nằm chung với gen của chuỗi α-globin trên nhiễm sắc thể, gồm chuỗi ζ và chuỗi α-globin, và nhóm nằm chung với gen của chuỗi β-globin trên nhiễm sắc thể, gồm các chuỗi ε, γ, β và δ. Các chuỗi globin xuất hiện tuần tự trong quá trình phát triển cá thể và sau khi cặp đôi với nhau sẽ tạo thành bốn loại chính Hb sau:
Hemoglobin “phôi”, hiện diện từ tuần thứ 3 đến tuần thứ 10 của thai kỳ và baogồm hai cặp đôi ζ2ε2, α2ε2 và ζ2γ2;
Hemoglobin “thai” (HbF α2γ2), tạo nên thành phần chính trong việc chuyên chở oxy trong thai kỳ/
Hemoglobin “người lớn” (HbA α2β2), thay thế HbF ngay sau khi sinh một thời gian ngắn và;
Một thành phần nhỏ hemoglobin người lớn khác là HbA2 (α2δ2).
Trong điều kiện bình thường, hồng cầu của người trưởng thành chứa 98% HbA, 2% HbA2 và vết HbF.
GEN GLOBIN VÀ SỰ TỔNG HỢP CHUỖI GLOBIN
Chuỗi globin có cấu trúc cực kỳ chính xác, đảm bảo cho khả năng chuyên tải oxy ngay lập tức từ phế nang của phổi và phân phối từ từ có kiểm soát cho mô. Cấu trúc chính xác của chuỗi globin được mã hóa bởi các gen nằm trên DNA của nhiễm sắc thể số 16 (nhóm chứa gen của chuỗi α) và số 11 (nhóm chứa gen của chuỗi β). Một số các nucleotide có vai trò “điều hòa” nằm dọc theo bên sườn của các gen cấu trúc, về phía trước (ở phía vị trí thứ 5’ của trình tự DNA, theo hướng “ngược dòng”) và tiếp theo sau đó (ở phía vị trí thứ 3’ của trình tự DNA, theo hướng “xuôi dòng”), nghĩa là chúng quyết định gen nào sẽ hoạt động hoặc không, cũng như mức độ hiệu quả hoạt động của các gen đó. Khi lớn lên, hầu hết sự tổng hợp globin xảy ra trong nguyên hồng cầu ở tủy xương. Hemoglobin phải có cấu trúc đúng và được sắp xếp sao cho số lượng chuỗi α bắt cặp chính xác với số lượng chuỗi β. Khi những điều kiện trên không được đáp ứng, hậu quả là sẽ gây khiếm khuyết một phần hay toàn bộ ở một hay cả hai gen “a-len” của chuỗi globin.
THALASSEMIA: ĐỊNH NGHĨA VÀ PHÂN BỐ TRÊN THẾ GIỚI
Thuật ngữ “thalassemia” liên quan đến một nhóm bệnh lý huyết học đặc trưng bởi sự giảm tổng hợp của một trong hai chuỗi polypeptide (α hoặc β) cấu tạo nên phân tử hemoglobin người lớn bình thường (HbA, α2β2), gây hậu quả giảm hemoglobin trong hồng cầu và thiếu máu.
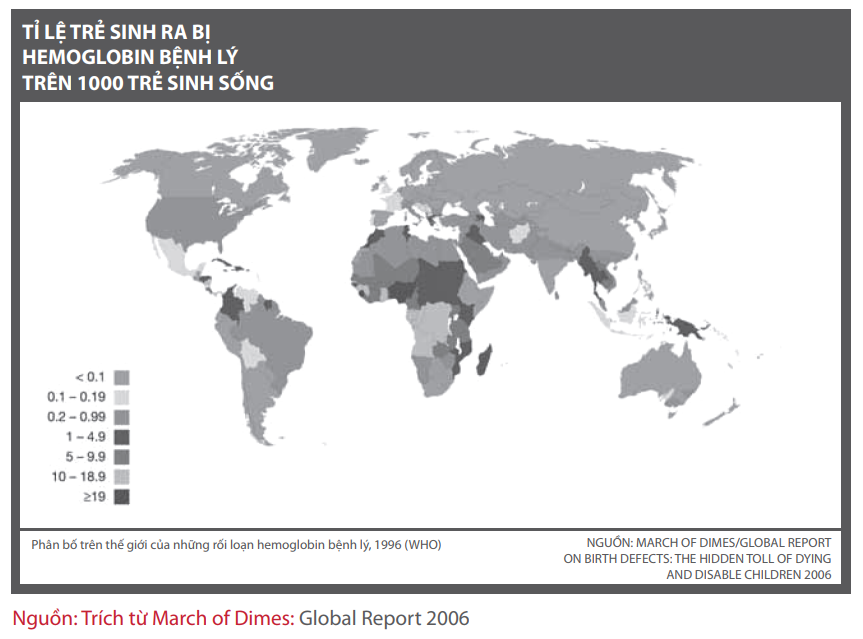
Tùy thuộc vào khiếm khuyết xảy ra trên gen nào và hậu quả tương ứng trên sự sản xuất các chuỗi globin mà hậu quả là sẽ gây ra bệnh α- hoặc β-thalassemia. Cuốn sách này chủ yếu đề cập đến nhóm bệnh β-thalassemia, mà hiện nay được công nhận là xảy ra khắp nơi trên thế giới vượt ra ngoài phạm vi những nước trước đây được xem là nguồn gốc lưu hành bệnh như những nước vùng Địa Trung Hải, Trung Đông, vùng xuyên biên giới Á-Âu, Ấn Độ đối với bệnh β-thalassemia và vùng Viễn Đông đối với bệnh α-thalassemia (Hình 1).
Β-THALASSAEMIA
Các thể bệnh
Theo quy luật, những cá thể dị hợp tử β-thalassemia (một gen a-len bị khiếm khuyết) có biểu hiện lượng Hb trung bình trong hồng cầu (MCH) giảm, thể tích trung bình hồng cầu (MCV) thấp, thay đổi hình thái nhẹ của hồng cầu, tỉ lệ HbA2 tăng, tỉ lệ sinh tổng hợp chuỗi β/α-globin giảm, và đôi khi kết hợp với tình trạng lượng Hb ở giới hạn thấp của bình thường hoặc hơi dưới mức bình thường. Trong những điều kiện bình thường, thalassemia thể ẩn không có liên quan đến bất kỳ ảnh hưởng lâm sàng quan trọng nào, chủ yếu vì hoạt động của gen β bình thường trên nhiễm sắc thể a-len tạo ra đủ lượng globin bền vững. Ngược lại, trường hợp di truyền mang hai gen của chuỗi β-globin khiếm khuyết gây ra nhiều thể bệnh lâm sàng khác nhau, từ mức độ lệ thuộc truyền máu (thalassemia thể nặng) đến thiếu máu nhẹ hoặc trung bình (thalassemia thể trung gian). Nghiên cứu về phân tử có thể phát hiện nhiều loại bất thường khác nhau và hỗ trợ vào việc chẩn đoán những thể bệnh nói trên.
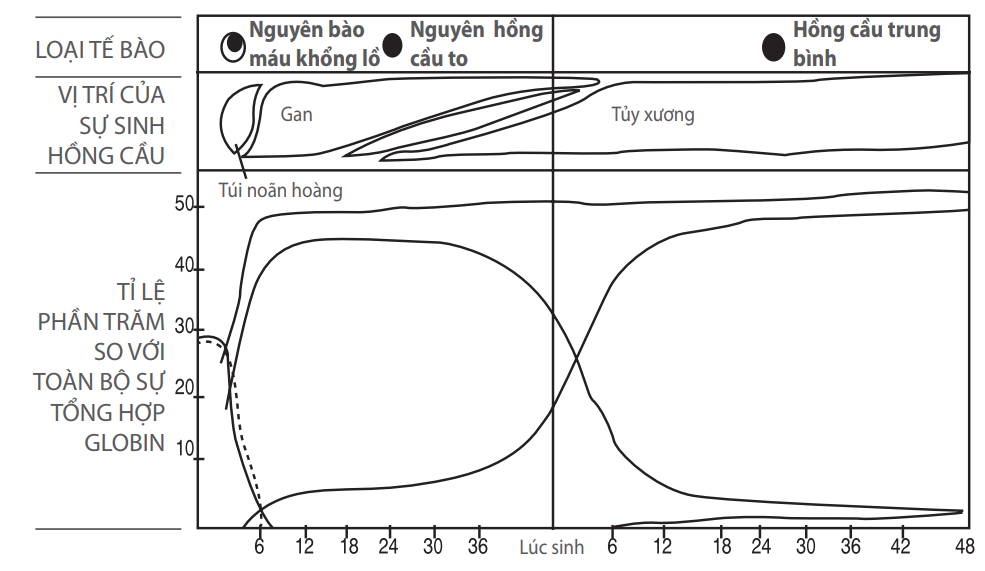
Hình 1: Sinh tổng hợp globin ở các giai đoạn phát triển khác nhau của phôi và thai
SINH LÝ BỆNH CỦA Β-THALASSEMIA
Những tiến bộ trong điều trị thalassemia đạt được chỉ sau khi sinh lý bệnh được làm sáng tỏ nhờ vào những nhà khoa học và y học. Hình 2 dưới đây phác họa sinh lý bệnh của β-thalassemia và mô tả chuỗi sự kiện xảy ra khi có sự mất cân bằng của chuỗi globin và tích tụ các chuỗi α-globin dư thừa, nghĩa là sinh hồng cầu không hiệu quả, thiếu máu, tủy xương nở rộng ra, biến dạng xương và tăng hấp thu sắt từ đường tiêu hóa.
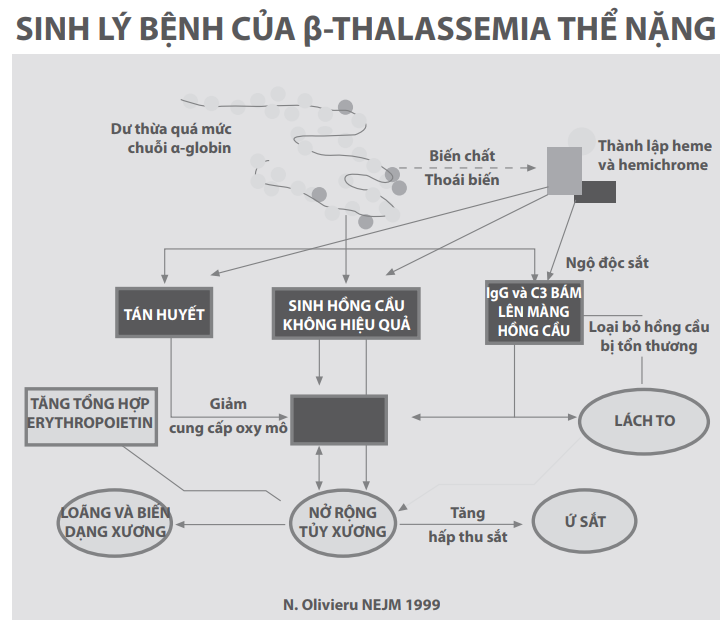
Hình 2: Hậu quả của sản xuất dư thừa chuỗi α-globin
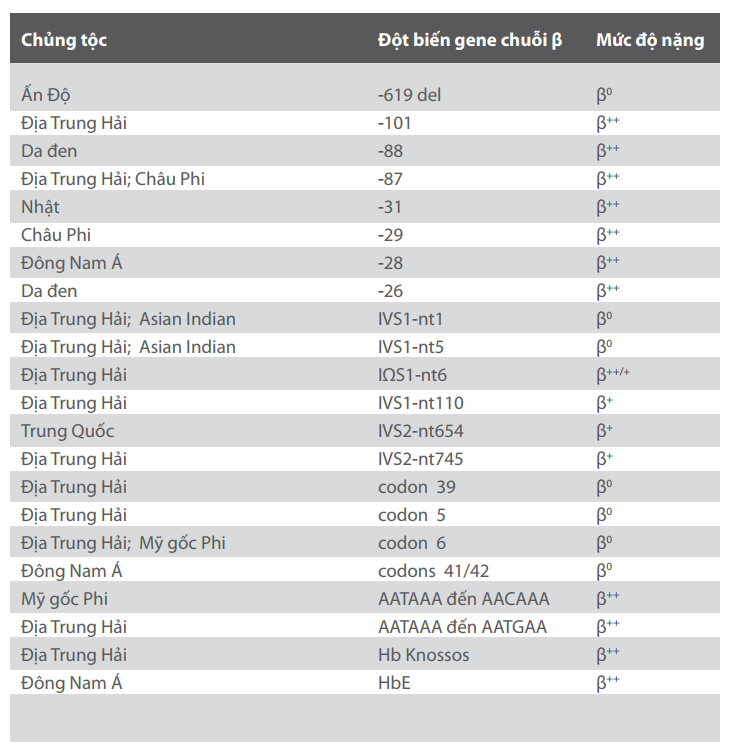
Mức độ mất cân bằng của chuỗi globin được quyết định bởi bản chất đột biến của gen chuỗi β. β0 là trường hợp hoàn toàn không còn sự tổng hợp chuỗi β-globin bởi các gen a-len khiếm khuyết. β+ là trường hợp các gen a-len còn sản xuất một phần chuỗi β-globin (khoảng 10%). Trong trường hợp β++, giảm tổng hợp chuỗi β-globin là rất nhẹ. Ngày nay đã có trên 200 loại đột biến thalassemia được ghi nhận. Bảng 1 bao gồm một số đột biến thalassemia thường gặp tùy theo phân bố chủng tộc cũng như mức độ nặng. Danh sách đầy đủ hơn về đột biến của β- thalassemia có thể được tìm thấy trên internet tại địa chỉ http://globin.cse.psu.edu/globin/html/huisman.
Những hemoglobin có cấu trúc chuỗi β-globin biến đổi liên quan đến điều trị thalassemia
Hemoglobin E là trường hợp rối loạn thay đổi cấu trúc thường gặp nhất có biểu hiện gần giống với bệnh thalassemia (xem chương 11: Thalassemia thể trung gian). HbE xảy ra do sự đột biến (G→A) ở vị trí 26 của gene chuỗi β-globin làm cho acid glutamic bị thay bởi acid lysin, hậu quả là khiếm khuyết gene chuỗi β-globin về số lượng lẫn chất lượng vì đột biến này liên quan đến sự kích hoạt của vị trí cắt đoạn tại đơn vị chứa mã thứ 24, 25, dẫn đến quá trình tạo chuỗi bị thay đổi. Hậu quả sau cùng là sự sản xuất lượng hemoglobin thay thế bị giảm đi (HbE).
HbE là hemoglobin bất thường phổ biến nhất ở khu vực Đông Nam Á, với tần suất mang bệnh lên đến trên 50% ở một số nơi. HbE cũng chiếm ưu thế ở một số vùng thuộc bán đảo Ấn Độ, bao gồm cả Bangladesh. Những trường hợp dị hợp tử HbE có lâm sàng bình thường và hiện diện 25-30% HbE trên điện di Hb cùng với thay đổi nhẹ các chỉ số hồng cầu. Thể đồng hợp tử HbE thường triệu chứng lâm sàng không rõ, có thể chỉ thiếu máu nhẹ. Khi khảo sát dưới kính hiển vi, phết máu ngoại biên cho thấy hồng cầu nhỏ với những hồng cầu hình bia chiếm 20-80%. Điện di Hb cho thấy 85-95% HbE và 5-10% HbF.
HbE/β-thalassemia là sự kết hợp phổ biến nhất của β-thalassemia với một hemoglobin cấu trúc bất thường, chiếm nhiều đa số ở vùng Đông Nam Á. Triệu chứng lâm sàng thay đổi tùy thuộc mức độ nặng – từ những trường hợp giống thalassemia thể trung gian đến những trường hợp thalassemia thể nặng lệ thuộc truyền máu. Nguyên nhân của sự khác nhau này chỉ được xác định một phần, vì những trường hợp có vẻ giống nhau về kiểu gene lại có biểu hiện lâm sàng rất khác nhau về mức độ nặng.
Hb Lepore là một bất thường cấu trúc khác của chuỗi β với sự kết hợp của gene chuỗi δ và chuỗi β. Đồng hợp tử của hemoglobin Lepore có thể gây ra hội chứng β-thalassemia lệ thuộc vào truyền máu từ mức độ nhẹ đến nặng.
Những rối loạn của Hemoglobin S: HbS, một hemoglobin bất thường phổ biến nhất trên thế giới, được hình thành do sự thay thế acid glutamic ở vị trí thứ 6 trên chuỗi β-globin bởi acid valine. Sự tương tác của β-thalassemia với HbS gây ra hội chứng rất giống với những rối loạn của bệnh hồng cầu hình liềm mà điển hình là không cần truyền máu suốt đời và do đó không bị tình trạng ứ sắt. Cũng như thalassemia, hướng dẫn về điều trị bệnh hồng cầu hình liềm đã được thành lập trong những năm gần đây và địa chỉ hữu ích để có thêm thông tin là: http://www.nhlbi.nih.gov/health/prof/ blood/sickle/sick-mt.htm.
α-thalassaemia
α-thalassemia là những rối loạn di truyền được đặc trưng bởi quá trình sản xuất chuỗi α-globin bị giảm hoặc ức chế. Các gen chuỗi α-globin ghép thành cặp và nằm ở điểm tận cùng của nhánh ngắn nhiễm sắc thể số 16. α-thalassemia gây ra thường nhất là thiếu hụt phần lớn những đoạn DNA chứa một hoặc cả hai gen chuỗi α-globin.
Tình trạng người lành mang gen không triệu chứng: Thiếu hụt một gen chuỗi α-globin gây ra tình trạng người lành mang gen không triệu chứng xảy ra rộng khắp trên thế giới.
α-thalassemia thể ẩn được đặc trưng bởi sự hiện diện còn sót lại của hai gen chuỗi α-globin còn hoạt động và không có liên quan đến bất kỳ biểu hiện trầm trọng về lâm sàng hoặc cận lâm sàng nào:
Thiếu máu mức độ trung bình và hồng cầu nhỏ nhược sắc.
Thiếu hoặc bất thường của ba gen chuỗi globin gây ra bệnh HbH, thường đặc trưng bởi thiếu máu mức độ trung bình, lách to và những cơn thiếu máu tán huyết cấp, chủ yếu do phản ứng với các loại thuốc có tính oxy hóa hoặc nhiễm trùng.
Những bất thường cấu trúc có liên quan khác mang tên hemoglobin Constant Spring, được đặc trưng bởi sự tổng hợp chuỗi α-globin không hiệu quả, do khiếm khuyết của một gen liên quan gây ra hậu quả kéo dài chuỗi globin. Đột biến này gặp chủ yếu ở Châu Á và sự di truyền đồng thời với thiếu hụt hai gen chuỗi α-globin sẽ gây ra thể nặng của bệnh HbH.
Phù nhau thai Hb Bart, biểu hiện lâm sàng nặng nhất của α-thalassemia, có đặc điểm chung là thiếu hụt tất cả bốn gen của chuỗi α-globin và thai chết trong tử cung. Thiếu hụt các gen chuỗi α-globin ở vị trí “cis” trên cùng một nhiễm sắc thể (α0-thalassemia) gặp phổ biến ở vùng Đông Nam Á nhưng lại hiếm gặp ở vùng Địa Trung Hải và ở Châu Phi thì càng hiếm hơn nữa.
Bệnh viện Nguyễn Tri Phương - Đa khoa Hạng I Thành phố Hồ Chí Minh
BÀI VIẾT KHÁC


