

️ Hội chứng rối loạn sinh tuỷ(Myelodysplastic syndrome) (P2)
PHÂN LOẠI MDS
Phân loại MDS được nhóm nghiên cứu Pháp - Mỹ - Anh đưa ra từ 1986 và gần đây (2001)TỔ chức Y tế thế giới (WHO) đưa ra cách phân loại theo mã số bệnh tật nói chung.
Phân loại theo FAB:
Cách phân loại này gồm 5 nhóm nhỏ:
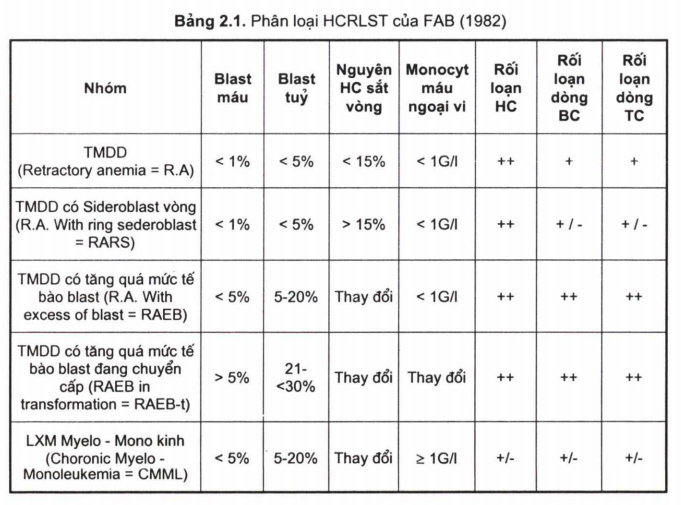
Phân loại trên đây của FAB đã giúp xếp các thể rối loạn sinh tuỷ vào 5 nhóm phục vụ cho công tác nghiên cứu các thể bệnh này. Tuy nhiên, phân loại này chưa gắn kết quả xét nghiệm với kết quả trên lâm sàng.
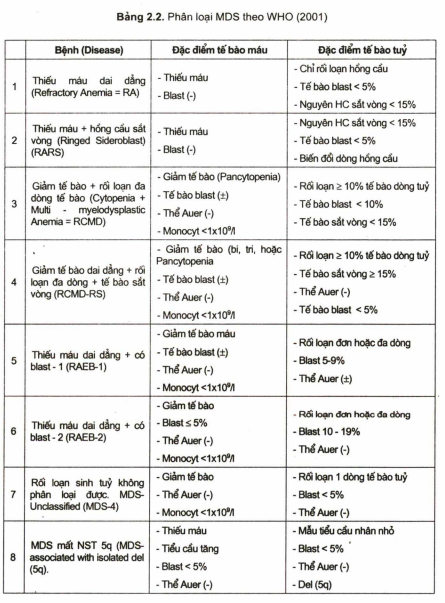
Phân loại của Tổ chức Y tế thế giới (WHO)
Gần đây (2001) WHO đưa ra cách phân loại bệnh quản lý theo mã số cho mỗi bệnh. Theo cách phân loại MDS được chia 8 thể (bảng 2.2); trong đó không có thể CMML như cách phân loại của FAB. Cách phân loại này giúp cho thầy thuốc tiên lượng và điều trị thuận lợi hơn.
Chẩn đoán xác định
Chẩn đoán xác định HCRLST chỉ có được sau khi nghiên cứu cẩn thận và đầy đủ tiêu bản máu ngoại vi, tuỷ xương và sinh thiết tuỷ, có hai tiêu chuẩn để chẩn đoán như sau:
Giảm một hoặc hai hoặc ba dòng tế bào ở máu ngoại vi, kết hợp với một tuỷ xương giàu tế bào hoặc tê bào tuỷ bình thường.
Rối loạn về hình thái xảy ra ở ít nhất một dòng tế bào, trong khi đó tăng tế bào non là bất thường có ý nghĩa lớn nhất.
Cần phải đánh giá mức độ của những rối loạn về hình thái: phải có ít nhất 10% tế bào của dòng đó có rối loạn về hình thái.
Chẩn đoán HCRLST nguyên phát là một chẩn đoán loại trừ.
Loại trừ các bệnh máu có kèm theo tình trạng loạn sản tuỷ (dysplasia)
Lơ xê mi cấp nguyên phát có kèm theo rối loạn nặng về hình thái. Trước tiên phải loại trừ mọi dữ kiện hướng đến sự chuyển từ HCRLST thành LXM cấp. Tỷ lệ blast trong tuỷ là luôn luôn trên 30%. Những trường hợp này thường kháng với điều trị tấn công, tỷ lệ lui bệnh thấp.
Lơ xê mi cấp thể M6: vấn đề đặt ra là khi tỷ lệ erythroblast là rất cao trong tuỷ (từ 50% trở lên) thì cần phân biệt thể RAEB - t hay LXM cấp thể M6. Trong trường hợp này cần xác định chính xác tỷ lệ blast. Ví dụ tỷ lệ erythroblast là 80%, myelobast là 12% trong tuỷ thì tỷ lệ blast so với 20% tế bào không thuộc dòng hồng cầu là 12/20 bằng 60%, vậy trường hợp này phải xếp là LXM cấp M6.
Hội chứng tăng sinh tuỷ: Đặc điểm chủ yếu của HCRLST là tình trạng loạn sản (dysplassia) và sinh máu không hiệu lực (ineffetive hematopoiesis) với mật độ tế bào tuỷ bình thường (normocclìulair) hoặc tăng (liypcrcellulair). Ngược lại hội chứng tăng sinh tuỷ (HCTST) hình ảnh tạo máu có hiệu lực với hình thái bình thường hoặc gần bình thường tuỷ rất giàu tế bào.
Loại trừ các bệnh lý khác có kèm theo tình trạng rối loạn sinh tuỷ:
Nhiễm tia xạ, hoá chất.
Ngộ độc kim loại nặng.
Ngộ độc rượu
Bệnh ung thư, nhiễm HIV.
Thiếu vitamin B12, acid folic.
Một số thể đặc biệt của HCRLST
HCRLST thứ phát sau điều trị hoá chất, tia xạ
Bệnh nhân ở mọi lứa tuổi sau một thòi gian điều trị bằng hoá chất tia xạ (thường sau 3-6 năm) sẽ có nguy cơ chuyển thành HCRLST hoặc LXM cấp. Nhóm alkylan là tác nhân gây HCRLST và LXM cấp cao hon các nhóm khác.
HCRLST thường dễ phát hiện do bệnh nhân đã được theo dõi từ trước.
Về huyết học thường có giảm cả ba dòng tế bào ở máu ngoại vi, tuỷ xương nghèo tế bào, xơ hoá mạnh kết hợp với rối loạn nặng về hình thái cấu trúc của cả ba dòng.
Về di truyền tế bào tỷ lệ tổn thương NST cũng cao hơn (> 80%) nhóm HCRLST nguyên phát. Hay gặp tổn thương trên NST số 5, 7.
Hội chứng rối loạn sinh tuỷ thể giảm tế bào tuỷ
Thể này chiếm 10 - 15% trường họp HCRLST nguyên phát.
Thường gặp ở người cao tuổi.
Về tế bào học cũng như thể điển hình, có giảm ba dòng máu ngoại vi nhưng tuỷ nghèo tế bào và một tiêu chuẩn rất quan trọng là có tình trạng rối loạn sinh máu thường là trên cả ba dòng tê bào.
Sinh thiết tuỷ xương là bắt buộc để khẳng định tình trạng giảm sinh tuỷ: khi mật độ tế bào tuỷ là < 30% ở người < 60 tuổi, và < 20% ở người > 60 tuổi.
Chẩn đoán phân biệt với suy tuỷ: dựa vào tình trạng rối loạn hình thái tế bào, đặc biệt là sự có mặt của mẫu tiểu cầu với hình thái bị rối loạn, ALIP, các đảo hồng cầu và xơ trên tiêu bản sinh thiết. Nếu có tổn thương về nhiễm sắc thể thì rất có giá trị.
Hội chứng rối loạn sinh tuý thế xơ tuỷ
Hầu hết các trương họp HCRLST nguyên phát đều có hiện tượng xo hoá tuỷ ở mức độ vừa và nhẹ. Tuy nhiên, có khoảng 11-15% trường họp có xớ hoá mạnh. Thể này thường có đặc điểm: giảm ba dòng tế bào ở máu ngoại vi (pancytopenia) hiếm khi có gan lách to, loạn sản 3 dòng, tăng tế bào tuỷ xương kèm theo có xơ phát triển mạnh có tăng sinh mẫu tiểu cầu đặc biệt là hình thái mẫu tiểu cầu nhỏ nhân thiểu thuỳ.
Chẩn đoán phân biệt với những bệnh lý khác có kèm theo xo hoá tuỷ: hội chứng tăng sinh tuỷ, LXM cấp thể M7... những bệnh lý này thường có đặc điểm: lách rất to, có hồng cầu non và bạch cầu non ra máu (leukoerythroblastosis), sinh máu ngoài tuỷ (extramedullary hematopoiesis), tăng sinh ba dòng tế bào mà không kèm theo loạn sản.
Hội chúng rối loạn sinh tuỷ sớm
Đó là các trường hợp: hồng cầu to (macrocytose) dai dẳng không kèm theo thiếu máu, tăng monocyt hoặc hiện tượng loạn sản, chỉ thấy ở một dòng tế bào ở mức độ nhẹ... Trường hợp này phải ấp dụng các nghiên cứu về di truyền, di truyền phân tử để phát hiện bất thưòng có tính chất clon. Trong nhiều trường hợp cần phải có thòi gian theo dõi để có thêm triệu chứng, giúp cho chẩn đoán.
TIẾN TRIỂN VÀ TIÊN LƯỢNG
Tiến triển
Tiến triển là mạn tính tất yếu dẫn đến tử vong do chuyển thành LXM cấp hoặc do nhiễm khuẩn, chảy máu, biến chứng của giảm tế bào máu hoặc ứ sắt là các biến chứng do truyền máu nhiều lần.
Theo phân loại FAB nhóm RA và RARS có đời sống trung bình là 3 năm với tỷ lệ chuyển LXM cấp < 15%, nhóm RAEB và CMML là 12 tháng và nhóm RAEB-t là 6 tháng với tỷ lệ chuyển thành LXM cấp là rất cao.
Các yếu tố tiên lượng trong HCRLST
Các thể theo phân loại FAB
Mức độ giảm tế bào ở giai đoạn chẩn đoán.
Tăng monocyt trong CMML.
Tỷ lệ blast ở máu và tuỷ
Bất thường về NST
Sự có mặt của ALIP
Nuôi cấy tuỷ
Dưới đây là cách tính điểm tiên lượng của Bournemouth. Mỗi một tiêu chuẩn cho 1 điểm:
Hb < 100 g/1
Bạch cầu trung tính < 2,5 GA hoặc > 15 GA.
Tiểu cầu < 100 GA
Blast tuỷ < 5%
Score từ 0-4. Người ta chia 3 nhóm tiên lượng:
Nhóm A = score 0-1 đời sống là 62 tháng;
Nhóm B = score 2-3 có đời sông 22 tháng
Nhóm C = score 3-4 có đời sống 8,5 tháng. Trong nhóm B đời sống của nhóm ALIP (-) là 34 tháng và AL1P (+) là 16 tháng.
ĐIỂU TRỊ
Điều trị triệu chứng
Truyền khôi hồng cầu chỉ đặt ra khi thiếu máu không thích nghi, cần hạn chế tôi đa nguy cơ lâu dài là các tai biến của truyền máu nhiều lần và ứ sắt.
Truyền tiểu cầu chỉ có chỉ định khi có chảy máu hoặc dự phòng chảy máu khi làm thủ thuật ngoại khoa. Cũng có nhiều nguy cơ do truyền máu nhiều lần.
Điều trị biến chứng nhiễm khuẩn: cần phải dùng kháng sinh sớm, phổ rộng cho tất cả hội chứng sốt.
Một số phương pháp điều trị đã dùng
Pyridoxin
Thường dùng cho thể thiếu máu RARS.
Liều 150 - 200mg/ngày dùng trong 3 tháng.
Tỷ lệ đáp ứng 1-2%.
Hormon
Cocticoid: thường không có hiệu quả
Andozen: kết quả hạn chê
Hoá trị liệu tăng cường
Chỉ định cho nhóm RAEB, RAEB-t và CMML
Thường dùng cytosin - arabinosid và anthracyclin.
Tỷ lệ đáp ứng tuỳ tác giả từ 15 - 51%, thời gian lui bệnh trung bình 6-8 tháng một số đạt 36 tháng.
Dưới 50 tuổi thường đáp ứng tốt hơn.
Ghép tuỷ đồng loài
Chỉ định hạn chế vì thường là bệnh nhân lớn tuổi và cần có người cho HLA tương đồng.
Chỉ định với bệnh nhân trẻ tuổi và có tiên lượng xấu.. Kết quả ban đầu là đáng khích lệ vì có khả năng loại trừ clon không bình thường. Biến chứng của ghép đồng loài.
Tác nhân biệt hoá
Aracytin liều thấp: 5-10mg/m2, tiêm dưới da cứ 12 giờ/1 lần trong 2-3 tuần.
Acid retinoique: 20mg/ngày trong 3 tuần. Đáp ứng một phần trong thời gian ngắn ở 20-30% trường hợp.
1.2.3 dehydro VTM D3, interferon còn đang trong thử nghiệm.
Phối hợp các tác nhân biệt hoá.
Các yếu tố tăng trưởng:
Được đưa vào với hy vọng điều chỉnh sự giảm tế bào, kích thích lại những clon bị ức chế. Đó là GM-CSF, G-CSF, erythropoietin, IL3.
Thuốc hoá chất đường uống:
6MP, hydrea, etoposid thường dùng trong thể CMML.
Chiến lược điều trị HCRLST: còn rất khó.
Cần có sự cân nhắc kỹ trước khi quyết định một phương pháp điều trị.
Tuổi cao, lâm sàng và xét nghiệm ổn định, tiến triển mạn tính chỉ cần theo dõi đơn thuần, điều trị triệu chứng khi cần thiết.
Tuổi trẻ, có nhiều yếu tố tiên lượng xâu, lâm sàng tiến triển, phải truyền máu nhiều lần, hay bị nhiễm khuẩn bắt buộc phải điều trị mạnh hơn.
Bảng 2.3. Liệt kê những rối loạn hình thái tê bào ở máu ngoại vi và tuỷ xương của
bệnh nhân bị hội chứng rối loạn sinh tuỷ (Checklist of morphological teatures of MDS)
|
Rối loạn sinh dòng hồng cầu (Dyserythropoiesis) |
Tuỷ xương (bone Marovv): Nhiều nhân nhỏ (multinuclearliy) Nhân vệ tinh (nuclear tragments) Nguyên hổng cầu khổng lổ (megaloblastoid changes) Bất thường về bào tương (cytoplassmic abnoimalities) Nguyên hổng cẩu sắt vòng (ringed sideroblasts) Tăng sinh hồng cầu (increased erythroblasts) Máu ngoại vi (peripheral blood): Hổng cầu nhiều hình thái (poikilocytosis) Hồng cầu to nhò (anisocytosis) Hồng cầu có chấm ưa base (bassophilic stippling) Hồng cầu có nhân ra máu ngoại vi (nucleated red blood cells) |
|
Rối loạn sinh dòng bạch cẩu hạt (Dysgranulopoiesis) |
Bất thường về nhân (nuclear abnormalitles including) Giảm đoạn (hypolobulation) Mầu nhân (nuclear sticks) Nhân dạng vòng (ring - shaped nuclei) Giảm hạt (hypogranulation) |
|
Rối loạn sinh dòng mẫu tiểu cẩu (Sysmegakaryopoiesis) |
Tuỷ xương (bone Marrow) Mẫu TC có kích thước nhỏ (micromegakaryocytes) Mẫu TC có 1 nhân lớn (large mononuclear forms) Mẫu TC có nhiều nhân nhỏ (multlple small nuclei) Giảm sinh mẫu TC (reduced numbers) Máu ngoại vi (peripheral blood) TC khổng lồ (large platelets). |
Bệnh viện Nguyễn Tri Phương - Đa khoa Hạng I Thành phố Hồ Chí Minh
BÀI VIẾT KHÁC


